Principles of Chemistry
Principles of Chemistry
 Projekcja Fischera i konfiguracja absolutna
Projekcja Fischera i konfiguracja absolutna
W celu dokładnego zrozumienia stereochemii przydatne jest zbadanie modeli cząsteczek. Nie jest to jednak możliwe podczas przedstawiania wzoru na papierze. W 1891 roku Emil Fischer opracował odwzorowanie Fischera, metodę przedstawiania czworościennych węgli na papierze. Wzór projekcyjny Fischera powstaje przez rzutowanie na płaszczyznę cząsteczki ustawionej pionowo do płaszczyzny rzutowania. Wtedy atom centralny znajduje się w płaszczyźnie papieru, atomy leżące powyżej i poniżej atomu centralnego znajdują się pod płaszczyzną papieru, atomy leżące z lewej i z prawej strony atomu centralnego znajdują się nad płaszczyzną papieru, a atom o najniższym lokancie znajduje się na górze. Stosując zapis wzorów w projekcji Fischera należy pamiętać, że są one rzutami na płaszczyznę. W tym przypadku dopuszczalny jest obrót wzoru o 180° ale już nie o 90°.
Dopuszczalne jest również ustalenie pozycji jednej grupy i obracanie pozostałych trzech w prawo lub w lewo. Jednak zamiana pozycji dowolnych dwóch grup powoduje konwersję enancjomeru do jego lustrzanego odbicia. Projekcje Fischera ograniczają się do cząsteczek których chiralność wynika z obecności asymetrycznych węgli (atomów). Natomiast w innych przypadkach należy użyć modelu przestrzennego cząsteczki aby stwierdzić jej chiralność.
Konfiguracja absolutna
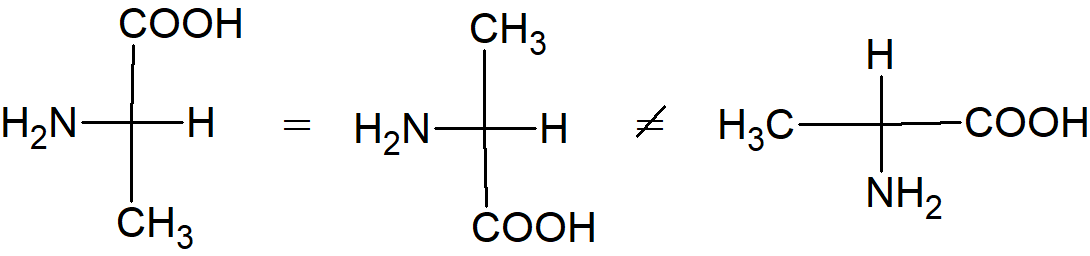
Przypuśćmy, że mamy dwie próbówki i w jednej z nich znajduje się kwas (–) mlekowy, a w drugiej (+) mlekowy. Nasuwa się pytanie, który enancjomer jest w której probówce. Oczywiście problem ten dotyczy dowolnych związków optycznie czynnych. A. Rosanoff zaproponował, aby wybrać jeden związek jako standard i arbitralnie przypisać mu konfigurację. Wybranym związkiem był jeden z izomerów aldehydu glicerynowego ze względu na jego pokrewieństwo z cukrami. Izomer (+) oznaczono symbolem D, a izomerowi (–) nadano symbol L.
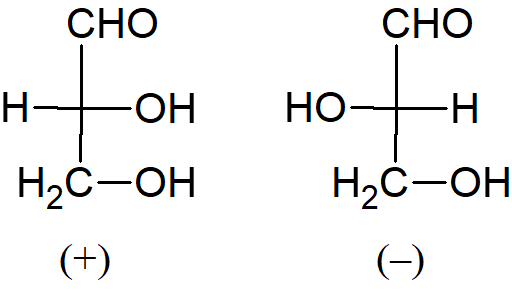
Po ustaleniu standardu można było na tej podstawie określać enancjomery innych związków. Na przykład aldehyd (+) glicerynowy, utleniony tlenkiem rtęci, daje kwas (–) glicerynowy. Ponieważ jest wysoce nieprawdopodobne, aby zmieniła się konfiguracja na centralnym atomie węgla, można wywnioskować, że kwas (–) glicerynowy ma taką samą konfigurację jak aldehyd (+) glicerynowy, a zatem kwas (–) glicerynowy jest również oznaczany symbolem D. Cząsteczki o tej samej konfiguracji nie muszą obracać płaszczyzny światła spolaryzowanego w tym samym kierunku. Fakt ten nie powinien nas dziwić, kiedy pamiętamy, że ten sam związek może obracać płaszczyznę światła spolaryzowanego w przeciwnych kierunkach w różnych warunkach. Gdy została ustalona konfiguracja kwasów glicerynowych (w odniesieniu do aldehydów glicerolowych), można było powiązać inne związki z ustalonym standardem Nawet związki bez asymetrycznych atomów, takie jak bifenyle i alleny, zostały umieszczone w szeregu D lub L. W momencie gdy związek zostaje zaszeregowany do klasy D lub L, mówi się, że znana jest jego konfiguracja absolutna. W 1951 roku stało się możliwe ustalenie, czy przypuszczenie Rosanoffa było słuszne. W oparciu o techniki krystalografii rentgenowskiej J. M. Bijvoet zbadał kryształ winianu sodowo rubidowego i stwierdził, że Rosanoff dokonał właściwego wyboru. Pomimo powszechnego użycia symboli D i L do oznaczenia konfiguracji absolutnej, metoda nie jest pozbawiona wad. Generalnie system D/L nie jest już używany, z wyjątkiem takich grup związków jak węglowodany i aminokwasy.
System, który zastąpił notację D/L, to system Cahna–Ingolda–Preloga, w którym cztery grupy asymetrycznego węgla są uszeregowane zgodnie z zestawem reguł sekwencyjnych. Dla naszych celów ograniczamy się tylko do kilku z tych reguł, które są wystarczające, aby poradzić sobie z ogromną większością związków chiralnych.
- Podstawniki są wymienione w kolejności malejącej liczby atomowej atomu bezpośrednio przyłączonego do węgla.
- Gdy dwa lub więcej atomów przyłączonych do asymetrycznego węgla jest takich samych, o kolejności decyduje liczba atomowa drugiego atomu. Na przykład w cząsteczce Me2CHCHBr CH2OH grupa CH2OH ma pierwszeństwo przed grupą Me2CH, ponieważ tlen ma wyższą liczbę atomową niż węgiel. Zauważmy, że tak jest, nawet jeśli są dwa węgle w Me2CH i tylko jeden tlen w CH2OH. Jeśli dwa lub więcej atomów połączonych z drugim atomem jest takich samych, to trzeci atom określa pierwszeństwo i tak dalej.
- Wszystkim atomom, z wyjątkiem wodoru, formalnie przypisano wartościowość 4. Tam, gdzie rzeczywista wartościowość jest mniejsza (jak w przypadku azotu, tlenu lub karboanionu), wprowadza się atomy fantomowe (oznaczone indeksem dolnym 0) używane do podniesienia wartościowości do czterech. Atomy fantomowe mają przypisaną liczbę atomową zero i z konieczności zajmują najniższe pozycje. Zatem ligand HNHMe2 plasuje się wyżej niż NMe2.
- Atom trytu ma pierwszeństwo przed deuterem, który z kolei ma pierwszeństwo przed zwykłym wodorem. Podobnie każdy wyższy izotop (np. 14C) ma pierwszeństwo przed każdym niższym.
- Wiązania podwójne i potrójne liczy się tak, jakby były podzielone, odpowiednio, na dwa lub trzy wiązania pojedyncze. Należy zauważyć, że w przypadku podwójnego wiązania C=C dwa atomy węgla są uważane za połączone z dwoma atomami węgla, a jeden z nich jest liczony jako mający trzy podstawniki pozorne.
Stosując powyższe zasady, wybrane grupy w malejącej kolejności to COOH, COPh, COMe, CHO, CH(OH)2, o-tolil, m-tolil, p-tolil, fenyl, C≡CH, tert-butyl, cykloheksyl, winyl, izopropyl, benzyl, neopentyl, allil, n-pentyl, etyl, metyl, deuter i wodór. Zatem cztery grupy aldehydu glicerynowego są ułożone w kolejności: OH, CHO, CH2OH, H. Po ustaleniu kolejności podstawników przy asymetrycznym atomie cząsteczka jest orientowana w taki sposób, że najniższa grupa w sekwencji jest skierowana z dala od patrzącego. Następnie, jeśli pozostałe grupy w określonej kolejności są zorientowane zgodnie z ruchem wskazówek zegara, cząsteczka jest oznaczona (R), a jeśli przeciwnie do ruchu wskazówek zegara, (S). W przypadku aldehydu glicerynowego enancjomer (+) to (R):
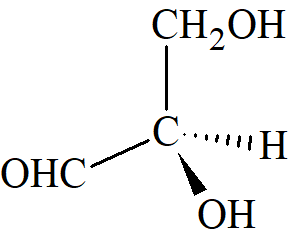
W większości przypadków system Cahna–Ingolda– Preloga jest jednoznaczny. To, czy mamy do czynienia z enancjomerem (R), czy (S) jest do określenia pod warunkiem że znamy konfigurację cząsteczki. System Cahna-Ingolda-Preloga został również rozszerzony na związki chiralne, które nie zawierają centrów stereogenicznych, ale mają symetrię chiralną. Związki posiadające taką symetrię to niesymetryczne alleny, biaryle wykazujące atropoizomerię. pochodne cykloheksanu, helicen, cyklofany, annuleny, trans-cykloalkeny i metaloceny.
Metody określania konfiguracji absolutnej
W niemal wszystkich metodach konieczne jest porównanie związku o nieznanej konfiguracji z takim, którego konfiguracja jest znana. Najważniejsze metody to:
Przekształcenie związku o nieznanej konfiguracji absolutnej (synteza związku o nieznanej konfiguracji) bez zakłócania centrum chiralności. Ponieważ centrum chiralności nie zostaje zakłócone, otrzymany związek ma oczywiście taką samą konfigurację jak związek wyjściowy. Nie musi to koniecznie oznaczać, że jeśli wyjściowy enancjomery jest typu (R), to konfiguracja końcowego produktu będzie również (R). Tak będzie, jeśli nie zostanie zakłócona sekwencja podstawników przy chiralnym atomie węgla. Na przykład podczas redukcji (R) -1-bromo-2-butanolu do 2-butanolu bez zmian w centrum chiralności, produktem jest izomer (S), ponieważ CH3CH2 plasuje się w hierarchii poniżej BrCH2, ale wyżej niż CH3.
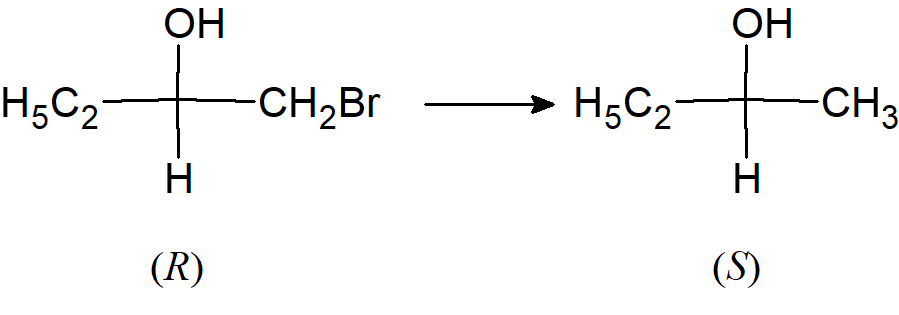
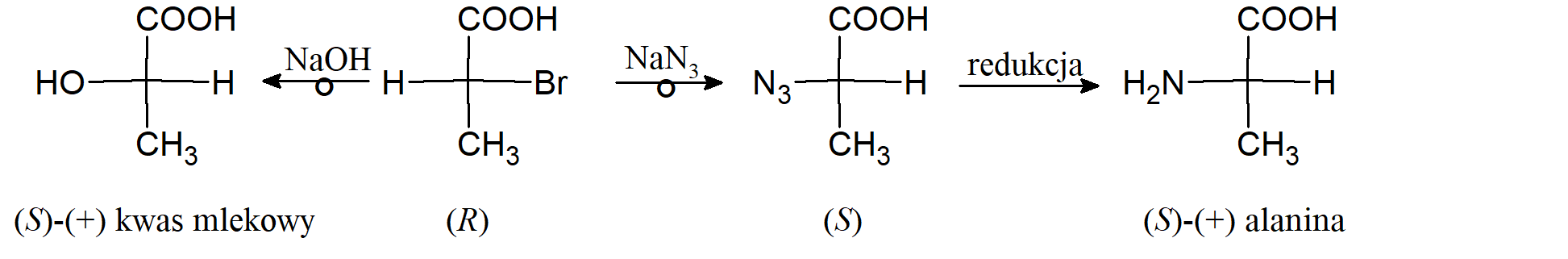
Konwersja w centrum chiralności, jeśli znany jest mechanizm zachodzących procesów. Mechanizm SN2 przebiega z odwróceniem konfiguracji na asymetrycznym węglu. Przeprowadzenie serii takich przekształceń pozwoliło stwierdzić, że kwas mlekowy ma taką samą konfigurację absolutną jak alanina.
Metody biochemiczne. W szeregu podobnych związków, takich jak aminokwasy czy pewne typy steroidów, dany enzym zazwyczaj oddziałuje tylko na cząsteczki o jednej, określonej konfiguracji. Jeśli enzym działa, powiedzmy, tylko formę L ośmiu aminokwasów, to jeżeli będzie działał na nieznany dziewiąty aminokwas to również on będzie miał formę L.
Porównanie z właściwościami optycznymi analogicznych związków. Czasami można użyć znaku i wielkości skręcalności aby określić z jakim izomer mamy do czynienia. W szeregu homologicznym skręcalność zwykle zmienia się stopniowo w określonym kierunku. Jeśli znane są konfiguracje wystarczającej liczby członków szeregu, konfiguracje brakujących elementów można określić metodą ekstrapolacji. Również niektóre podstawniki zmieniają w mniejszym lub większym stopniu skręcalność cząsteczki wyjściowej, zwłaszcza gdy jest ona układem sztywnym, takim jak steroid.
Jedną z najbardziej przydatnych metod określania składu enancjomerycznego jest derywatyzacja alkoholu chiralnym odczynnikiem nieracemicznym i badanie proporcji otrzymanych diastereomerów metodą chromatografii gazowej. Istnieje wiele dostępnych środków derywatyzujących, ale najczęściej stosowane są pochodne kwasu a-metoksy-a-trifluorometylofenylooctowego. Reakcja z chiralnym alkoholem nieracemicznym (R*OH, gdzie R* jest grupą zawierającą centrum stereogeniczne) generuje ester Moshera, który można analizować pod kątem składu diastereomerycznego za pomocą 1H lub 19F NMR, jak również technikami chromatograficznymi. Alternatywnie można dokonać kompleksowania z lantanowcami co pozwala na rozdzielenie sygnałów estru i wykorzystanie ich do określenia składu enancjomerycznego.
-3,3,3-trifluoro-2-metoksy-2-fenylopropanowy") kwas (2RS)-3,3,3-trifluoro-2-metoksy-2-fenylopropanowy
kwas (2RS)-3,3,3-trifluoro-2-metoksy-2-fenylopropanowy
Inne metody opierają się na syntezie asymetrycznej czy też badaniach dichroizmu kołowego lub optycznej dyspersji skręcalności.
Cząsteczki z więcej niż jednym centrum stereogenicznym
Gdy cząsteczka ma dwa centra stereogeniczne, każde z nich posiada swoją własną konfigurację, którą można sklasyfikować jako (R) lub (S) zgodnie z metodą Cahna–Ingolda–Preloga. W sumie występują cztery izomery, ponieważ każde centrum może być typu (R) lub (S). Ponieważ dla cząsteczki istnieje tylko jedno lustrzane odbicie, tylko jeden z pozostałych trzech izomerów może być enancjomerem A. W przypadku przedstawionym na schemacie jest to B; przy odbiciu lustrzanym (R) przechodzi zawsze w (S). Zarówno C jak i D są drugą parą enancjomerów.
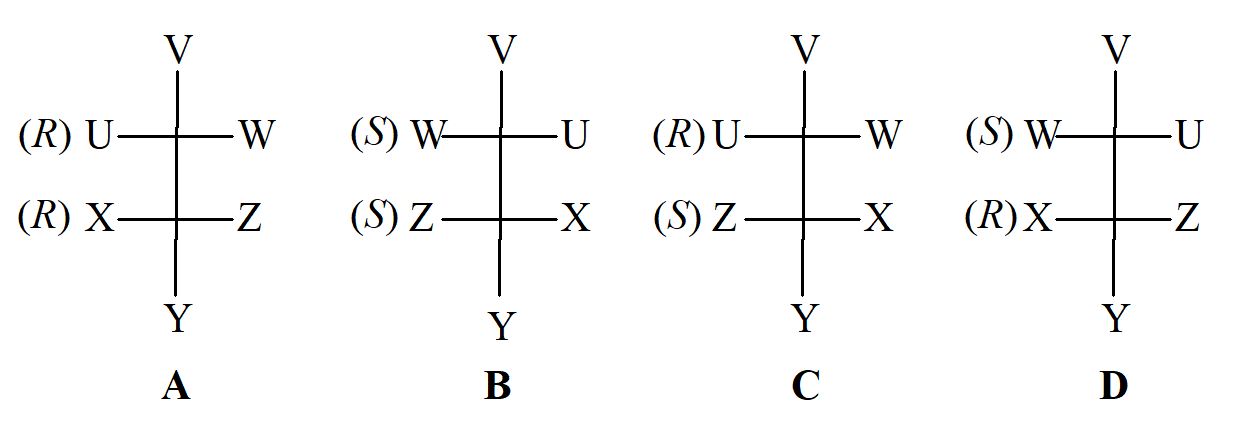
Różnica pomiędzy C i D w stosunku do A i B określana jest terminem diastereoizomerii. Diastereoizomery można zdefiniować jako stereoizomery, które nie są enancjomerami. Ponieważ C i D są enancjomerami, muszą mieć identyczne właściwości (z pewnymi wyjątkami) i to samo dotyczy A i B. Jednakże właściwości A i B nie są identyczne z właściwościami C i D. Mają one różne temperatury topnienia, wrzenia, rozpuszczalności, reaktywność i wszystkie inne właściwości fizyczne, chemiczne i spektralne. Właściwości te są zwykle podobne, ale nie identyczne. W szczególności diastereoizomery mają różne wartości skręcalności właściwej; w rzeczywistości jeden diastereoizomer może być chiralny i obracać płaszczyznę światła spolaryzowanego, podczas gdy inny może być achiralny. W związku z tym staje się jasne dlaczego enancjomery reagują z różną szybkością z innymi chiralnymi cząsteczkami, ale w tym samym tempie z cząsteczkami achiralnymi. W tym drugim przypadku kompleks aktywny utworzony z (R) enancjomeru i cząsteczki drugiego reagenta jest lustrzanym odbiciem kompleksu aktywnego utworzonego z enancjomeru (S) i cząsteczki reagenta. Ponieważ dwa kompleksy aktywne są enancjomeryczne, ich energie są takie same, a szybkości reakcji, w których powstają, muszą być takie same. Jednak gdy enancjomer (R) reaguje z chiralną cząsteczką, która ma, powiedzmy, konfigurację (R), kompleks aktywny ma dwa centra chiralne o konfiguracjach (R) i (R), podczas gdy kompleks aktywny utworzony z enancjomery (S) ma konfiguracje (S)(R). Oba kompleksy aktywne są diastereomerami, nie mając takich samych energii i w konsekwencji tworzą się z różną szybkością.
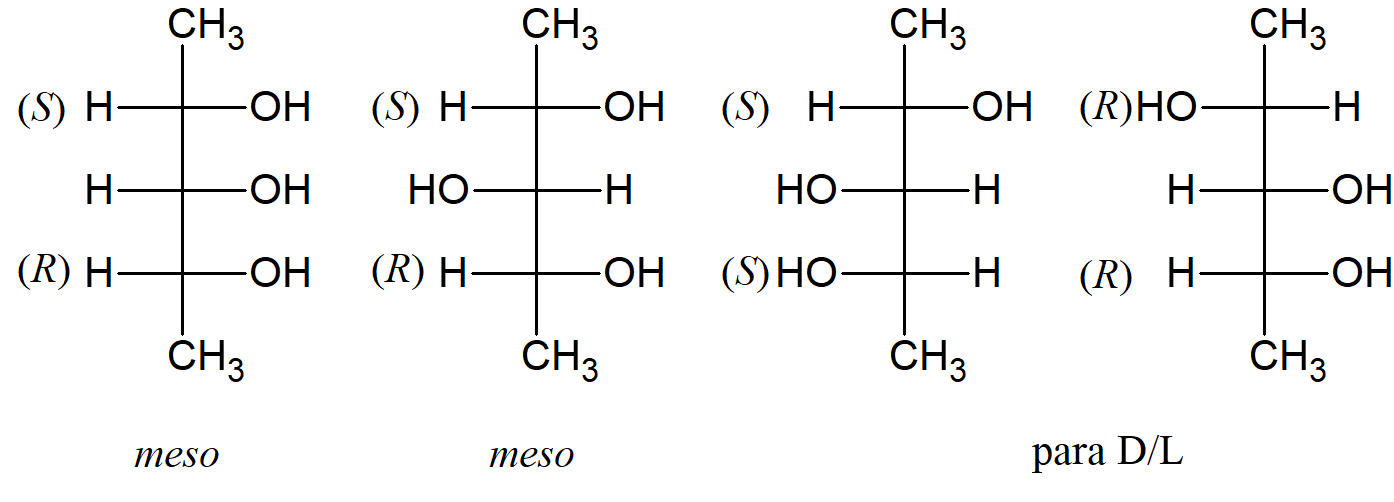
Chociaż cztery to maksymalna możliwa liczba izomerów, gdy związek ma dwa centra stereogeniczne (związki chiralne bez chiralnego węgla lub z jednym chiralnym atomem węgla i innym typem centrum stereogenicznego również podlegają opisanym tutaj regułom), niektóre związki mają ich mniej. Kiedy trzy grupy na jednym chiralnym atomie są takie same jak na drugim, jeden z izomerów (zwany formą mezo) ma płaszczyznę symetrii, a zatem jest optycznie nieaktywny, mimo że posiada dwa chiralne atomy węgla. Kwas winowy to typowy przypadek. Istnieją tylko trzy izomery kwasu winowego: para enancjomerów i nieaktywna forma mezo. W przypadku związków, które mają dwa chiralne atomy, formy mezo występują tylko wtedy, gdy cztery podstawniki przy jednym z chiralnych atomów są takie same jak przy drugim atomie chiralnym.
 Trzy stereoizomery kwasu winowego
Trzy stereoizomery kwasu winowego
W większości przypadków z więcej niż dwoma centrami stereogenicznymi liczbę izomerów można obliczyć ze wzoru 2n, gdzie n jest liczbą centrów chiralnych, chociaż w niektórych przypadkach faktyczna liczba jest niższa, ze względu na formy mezo. Interesującym przypadkiem jest 2,3,4-pentanotriol. Środkowy węgiel nie jest asymetryczny, gdy obydwa węgle 2– i 4– to (R) (lub S), ale jest asymetryczny, gdy jeden z nich to (R), a drugi to (S). Taki węgiel nazywany jest węglem pseudoasymetrycznym. W takich przypadkach istnieją cztery izomery: dwie formy mezo i jedna para D/L. Dwa diastereoizomery, które mają różną konfigurację tylko w jednym centrum chiralności, nazywane są epimerami.
W przypadku związków z dwoma lub więcej centrami chiralności konfigurację bezwzględną należy określić oddzielnie dla każdego centrum. Procedura polega na określeniu konfiguracji dla jednego centrum, a następnie powiązaniu konfiguracji na tym atomie z innymi w cząsteczce. Jedną z metod jest krystalografia rentgenowska, której nie można zastosować do określenia konfiguracji absolutnej w konkretnym centrum stereogenicznym, ale która daje względne konfiguracje wszystkich centrów stereogenicznych w cząsteczce, a tym samym absolutną konfigurację wszystkich pod warunkiem, że dla jednego centrum zostanie ona określana niezależnie. Powstaje problem, jak nazwać różne stereoizomery związku, gdy jest ich więcej niż dwa. Enancjomery są praktycznie zawsze określane tą samą nazwą z rozróżnieniem przy użyciu (R) i (S) lub D i L lub (+) lub (–). We wczesnych etapach chemii organicznej zwyczajem było nadawanie każdej parze enancjomerów innej nazwy lub przynajmniej stosowanie przedrostków typu epi-, peri- itp. Tak więc aldoheksozy nazywane są glukozą, mannozą, i tak dalej, chociaż wszystkie są 2,3,4,5,6-pentahydroksyheksanalem (w ich formach o otwartym łańcuchu). Ta praktyka była częściowo spowodowana brakiem wiedzy na temat tego z jakimi konfiguracjami mamy do czynienia przy poszczególnych izomerach. Obecnie jest zwyczajowo opisywać każdą pozycję chiralną osobno jako (R) lub (S) lub używając innych symboli. Tak więc w przypadku steroidów grupy powyżej „płaszczyzny” układu pierścieni są oznaczone symbolem β, a te poniżej α. Linie ciągłe są często używane do przedstawiania grup β, a linie przerywane do grup α.
 1α-Chloro-5-cholesten-3β-ol
1α-Chloro-5-cholesten-3β-ol
W przypadku wielu związków o otwartym łańcuchu stosowane są przedrostki, które pochodzą od nazw odpowiednich cukrów i opisują raczej cały system niż każde centrum chiralności oddzielnie.
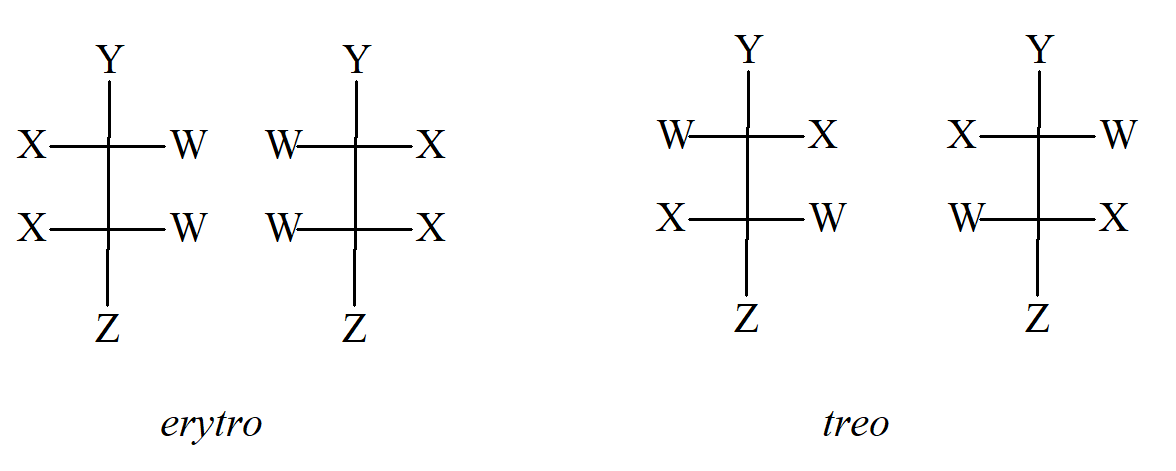
Dwa takie powszechnie używane przedrostki to erytro- i treo-, które są stosowane do układów zawierających dwa asymetryczne atomy węgla, gdy dwie z grup są takie same, a trzecia jest inna. Para erytro ma identyczne grupy po tej samej stronie, gdy jest rysowana zgodnie z konwencja Fischera, a gdyby Y zostało zmienione na Z, byłoby to mezo. Para treo ma je po przeciwnych stronach i gdyby Y zostało zmienione na Z, nadal byłaby to para D/L. Inny system oznaczania stereoizomerów używa terminów syn i anti. „Główny łańcuch” cząsteczki jest narysowany zwykłym zygzakiem. Następnie, jeżeli dwa podstawniki inne niż wodór znajdują się po tej samej stronie płaszczyzny określonej przez łańcuch główny, stosuje się przedrostek syn–; w przeciwnym razie mamy układ anti–.