 Principles of Chemistry
Principles of Chemistry
 Substytucja elektrofilowa
Substytucja elektrofilowa
Większość reakcji podstawienia na alifatycznym atomie węgla jest dokonywana przez nukleofile. W układach aromatycznych sytuacja jest odwrotna, ponieważ wysoka gęstość elektronów w pierścieniu aromatycznym prowadzi do jego reaktywności jako zasady Lewisa lub zasady Brønsteda-Lowry'ego. W przypadku podstawień elektrofilowych dodatni jon lub dodatni koniec dipola lub dipola indukowanego jest atakowany przez pierścień aromatyczny. Grupa opuszczająca musi zatem odejść pozbawiona pary elektronowej. W przypadku podstawień nukleofilowych głównymi grupami opuszczającymi są te, które są w stanie przenosić niezaangażowaną w wiązanie parę elektronową: Br–, H2O, OTs–, czyli słabe zasady. W podstawieniach elektrofilowych najważniejszymi grupami opuszczającymi są te, które mogą istnieć bez pary elektronów potrzebnych do wypełnienia powłoki zewnętrznej, czyli najsłabsze kwasy Lewisa.
Substytucje Elektrofilowe związków aromatycznych różnią się od substytucji nukleofilowych tym, że znaczna większość tych procesów przebiega według tylko jednego mechanizmu w odniesieniu do substratu. W tym mechanizmie atakowany jest elektrofil (który można postrzegać jako kwas Lewisa). przez elektrony π pierścienia aromatycznego (zachowujące się w większości przypadków jak zasada Lewisa) w pierwszym etapie. Ta reakcja prowadzi do powstania nowego wiązania C–X i nowego węgla o hybrydyzacji sp3 w dodatnio naładowanym półprodukcie (X jest elektrofilem). Dodatnio naładowany związek pośredni jest stabilizowany przez rezonans, ale nie jest związkiem aromatycznym. W momencie utraty protonu od węgla sp3, który „sąsiaduje” z dodatnim węglem w powstałym jonie arenowym powoduje odtworzenie aromatyczności pierścienia. Proton jest zatem grupą opuszczającą w ogólnej reakcji, w której X zastępuje H.
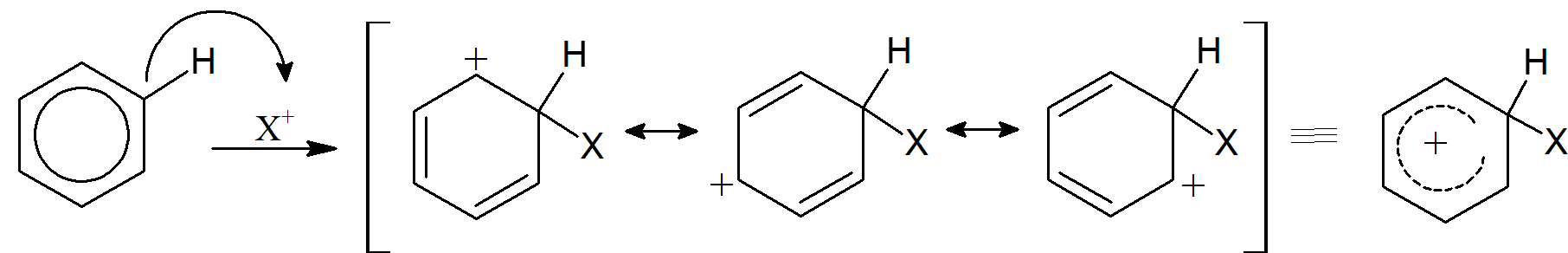
Elektrofil może być jonem dodatnim lub cząsteczką, która ma dodatni dipol. Jeśli jest to jon dodatni, zostaje zaatakowany przez pierścień (para elektronów z aromatycznego sekstetu jest przekazywana elektrofilowi), tworząc karbokation. Karbokationy mogą reagować na różne sposoby, ale w przypadku tego typu jonów najbardziej prawdopodobną drogą jest utrata X+ lub H+. W drugim etapie mechanizmu reakcja przebiega z utratą protonu, a aromatyczny sekstet elektronowy zostaje odtworzony w produkcie końcowym.
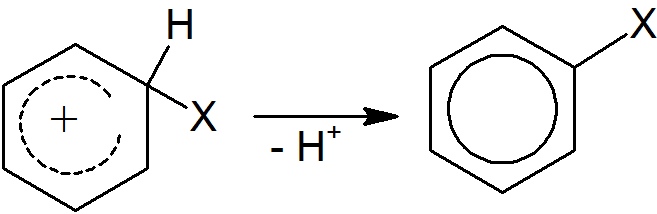
Drugi etap reakcji jest niemal zawsze szybszy niż pierwszy.
Jeśli atakujący związek nie jest jonem, ale dipolem, produkt musi mieć ładunek ujemny, chyba że część dipola wraz z parą elektronową zostanie oderwana na którymś z etapów procesu.
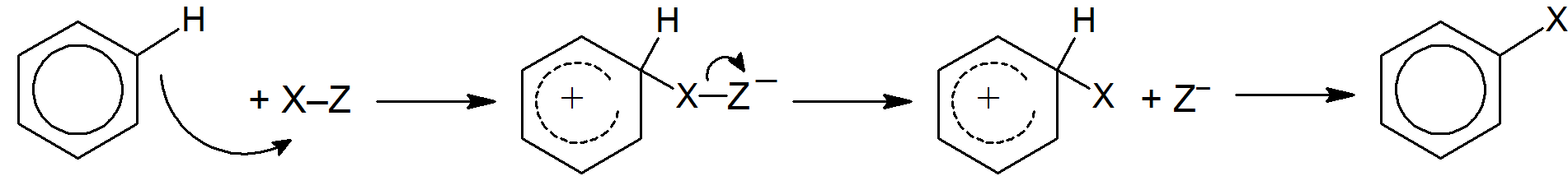
Mechanizm SE1 (jednocząsteczkowa substytucja elektrofilowa) jest rzadki, występuje tylko w niektórych przypadkach, w których atomem odchodzącym jest węgiel lub w obecności bardzo silnej zasady. Składa się on z dwóch etapów z pośrednim karboanionem.
Miejsce podstawienia i reaktywność monopodstawionych pierścieni benzenowych
Gdy reakcja podstawienia elektrofilowego jest przeprowadzana na monopodstawionym benzenie, nowa grupa może być kierowana do pozycji orto, meta lub para, a podstawienie może być wolniejsze lub szybsze niż w przypadku samego benzenu. Grupa obecna w pierścieniu benzenowym decyduje, jaką pozycję zajmie nowy podstawnik i czy reakcja będzie wolniejsza czy szybsza niż w przypadku benzenu. Grupy, które zwiększają szybkość reakcji, nazywane są aktywującymi, a te, które spowalniają dezaktywację. Niektóre grupy kierują podstawienie w pozycji meta i grupy te dezaktywują pierścień. Grupy kierujące podstawnik w pozycje orto i para również mogą dezaktywować pierścień ale większość z nich aktywuje układ benzenowy.
Efekty wywoływane przez obecne w pierścieniu benzenowym podstawniki można wyjaśnić rozpatrując rezonans i wpływu podstawnika na stabilność pośredniego jonu arenowego. Należy pamiętać, że przebieg tych reakcji jest kontrolowany kinetycznie, a nie termodynamicznie. Niektóre reakcje są nieodwracalne, a inne są zwykle zatrzymywane na długo przed osiągnięciem równowagi. Dlatego to, który z trzech możliwych związków pośrednich jest tworzony, nie zależy od termodynamicznej stabilności produktów, ale od energii aktywacji niezbędnej do utworzenia każdego z trzech produktów pośrednich. Nie jest łatwo przewidzieć, która z energii aktywacji ma najniższą wartość ale zakładamy, że energia stanu przejściowego jest zbliżona do energii jonu arenowego. Możemy zatem przyjąć, że geometria stanu przejściowego przypomina geometrię stanu przejściowego i że wszystko, co zwiększa stabilność stanu przejściowego, obniża również energię aktywacji niezbędną do jego osiągnięcia. Ponieważ po utworzeniu stan przejściowy szybko przekształca się w produkty, możemy wykorzystać względne stabilności trzech stanów przejściowych jako wytyczne do przewidywania, które produkty będą głównie powstawać. Trzy możliwe jony powstałe w wyniku przyłączenia Y w pozycjach orto, meta i para są pokazane na poniższym schemacie.
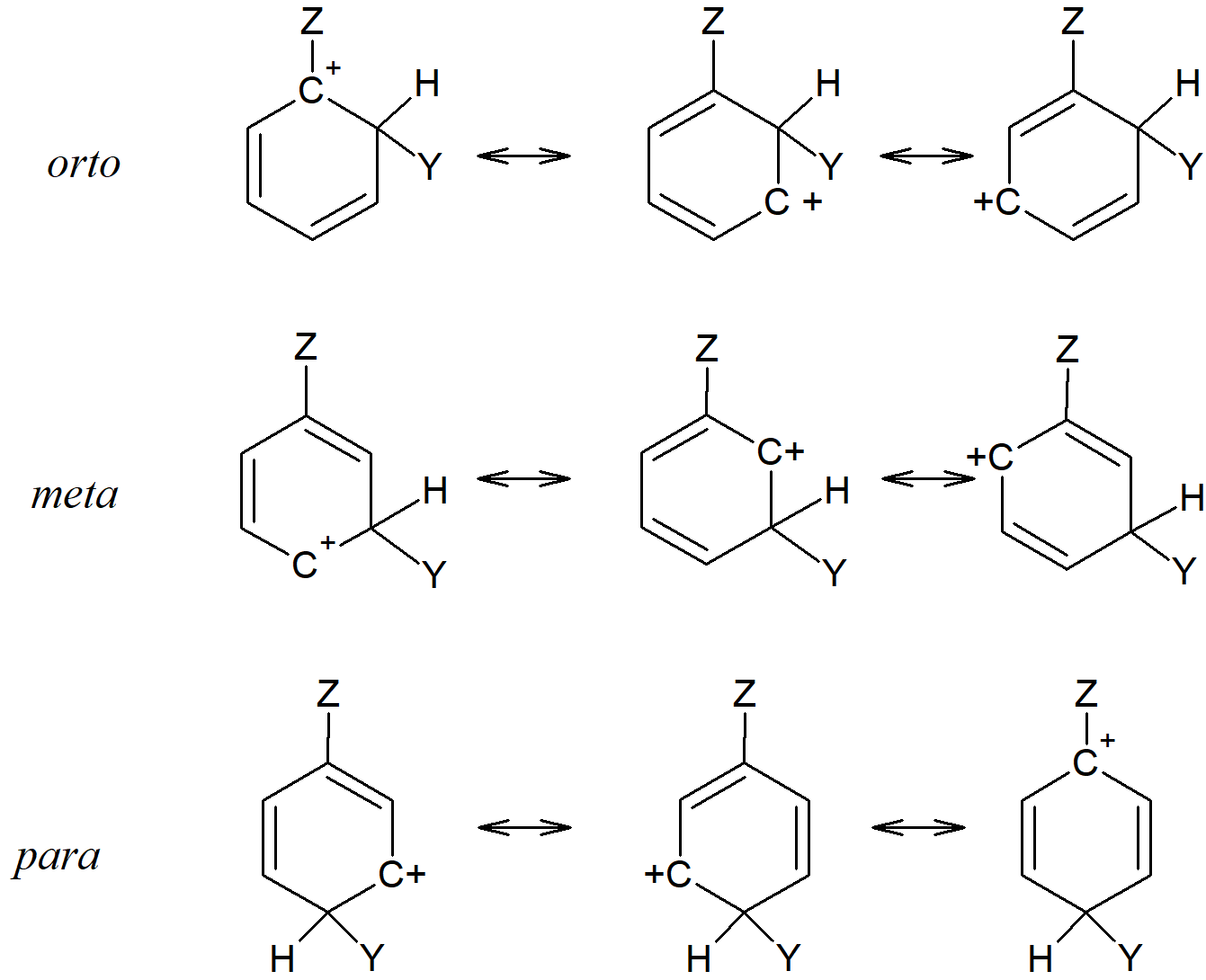
Możemy zatem przewidzieć, że każda elektrodonorowa grupa Z powinna ustabilizować wszystkie trzy formy jonów, ponieważ oddawanie elektronów do dodatniego centrum pierścienia stabilizuje go. Z drugiej strony grupy odciągające elektrony zwiększą dodatni ładunek na pierścieniu i destabilizują jon arenowy. Takie efekty powinny zmniejszać się wraz z odległością, a zatem są najsilniejsze przy węglu przyłączonym do grupy Z (węgiel ipso). Z trzech jonów arenowych, tylko orto i para mają ładunek dodatni na tym węglu. Żadna z kanonicznych form jonu meta nie ma ładunku dodatniego na węglu ipso. Dlatego grupy elektronodonorowe powinny stabilizować wszystkie trzy jony, ale przede wszystkim orto i para, a więc nie tylko aktywować, ale także kierować podstawniki w pozycje orto i para. Z drugiej strony grupy elektronoakcpetorowe powinny destabilizować wszystkie trzy jony, ale głównie orto i para, czyli powinny być nie tylko grupami dezaktywującymi pierścień, ale także kierującymi podstawnik w pozycję meta.
Niektóre podstawniki mają parę elektronów, które mogą wpływać na pierścień. W takim wypadku jony arenowe wyglądają następująco:
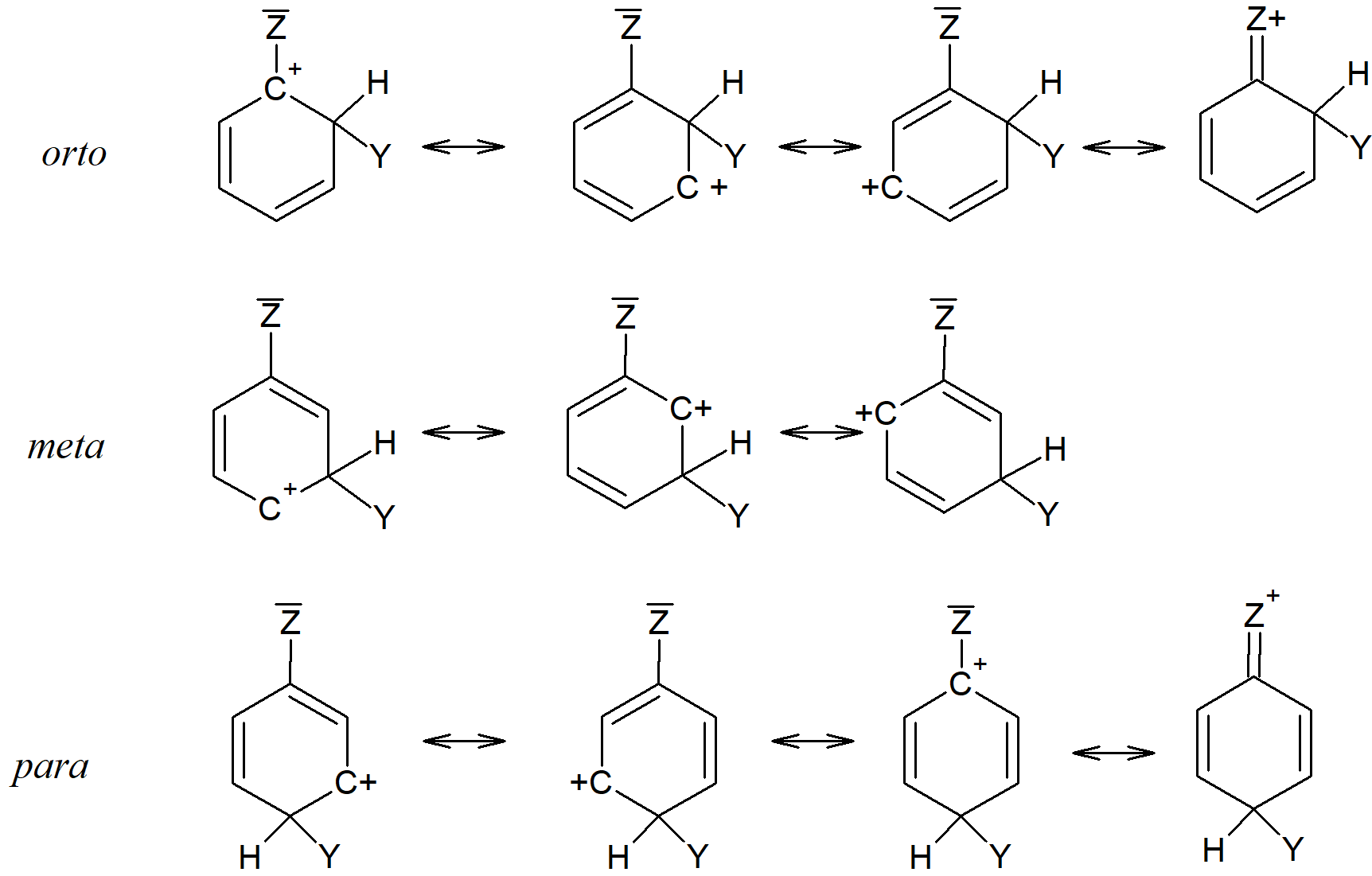
Dla każdego jonu można narysować te same trzy formy kanoniczne, jak poprzednio, ale teraz możemy narysować dodatkową postać dla jonów orto i para. Stabilność tych dwóch jonów jest zwiększona przez dodatkową formę nie tylko dlatego, że jest to inna forma kanoniczna, ale również dlatego, że jest bardziej stabilna niż inne i ma większy wkład w hybrydę. Każdy atom (oprócz oczywiście wodoru) w tych dwóch końcowych formach orto i para ma pełny oktet, podczas gdy wszystkie inne formy mają jeden atom węgla z sekstetem. Nie można narysować odpowiedniej formy dla izomeru meta. Włączenie tej formy do hybrydy obniża energię rozprzestrzeniając ładunek dodatni na grupę Z. Grupy z parą elektronów (np. halogeny) zatem nie tylko kierują podstawienie w pozycje orto i para, ale także aktywują te pozycje pod względem ataku elektrofilowego.
Można wyróżnić trzy typy podstawników w pierścieniu benzenowym.
- Grupy posiadające parę elektronów na atomie połączonym z pierścieniem. W tej kategorii znajdują się O–, NR2, NHR, NH2, OH, OR, NHCOR, OCOR, SR i cztery halogeny. Halogeny dezaktywują pierścień aromatyczny i szybkość reakcji jest wolniejsza niż w przypadku benzenu, efekt ten może wynikać z wyjątkowego poziomu energii orbitalu wolnej pary halogenów, który jest wyższy niż odpowiedni orbital π benzenu. Grupa SH prawdopodobnie też powinna wywoływać taki sam efekt, z tym wyjątkiem, że w przypadku tiofenoli elektrofile zwykle atakują raczej siarkę niż pierścień, a podstawienie pierścienia nie jest możliwe w przypadku tych związków. Rozpatrzenie struktur rezonansowych pozwala stwierdzić, że wszystkie te grupy powinny kierować podstawienie w pozycje orto i para. Silnie aktywującymi grupami są NR2, NHR, NH2 i OH, podobnie jak O–. Pozostałe grupy są łagodnie aktywujące, z wyjątkiem halogenów, które są dezaktywujące. Fluor jest najmniej dezaktywujący, a fluorobenzeny zwykle wykazują reaktywność zbliżoną do reaktywności samego benzenu. Pozostałe trzy halogeny dezaktywują pierścień w porównywalnym stopniu. Aby wyjaśnić, dlaczego chlor, brom i jod dezaktywują pierścień, mimo że kierują podstawniki w pozycje orto i para, należy założyć, że ostatnie formy kanoniczne mają tak duży wkład w hybrydy, że tworzą jony orto i para arenu bardziej stabilne niż meta, mimo że efekt destabilizujący halogenu powoduje wyciągnięcie gęstości elektronowej z pierścienia i jego dezaktywowanie. Cl, Br i J sprawiają, że jony orto i para są bardziej stabilne niż meta, ale mniej stabilne niż niepodstawiony jon arenowy. W przypadku innych grup, które zawierają wolną parę elektronową, jony orto i para są bardziej stabilne niż jon meta lub niepodstawiony jon benzenowy. Dla większości grup w tej kategorii jon meta jest bardziej stabilny niż jon benzenowy, więc grupy takie jak NH2 i OH również aktywują pozycje meta, ale w mniejszym stopniu niż pozycje orto i para.
- Grupy, które nie posiadają par elektronowych na atomie połączonym z pierścieniem, a są grupami dezaktywującymi. W przybliżonej kolejności malejącej zdolności dezaktywacji możemy wyróżnić tutaj NR3+, NO2, CF3, CN, SO3H, CHO, COR, COOH, COOR, CONH2, CCl3 i NH3+. Do tej kategorii należą również wszystkie inne grupy z dodatnim ładunkiem na atomie bezpośrednio połączonym z pierścieniem (SR2+, PR3+), oraz wiele grup z dodatnimi ładunkami na dalszych atomach. Wszystkie te podstawniki kierują podstawienie w pozycję meta i dezaktywują pierścień z wyjątkiem NH3+. Grupa NH3+ stanowi pewną anomalie kierując podstawnik w pozycję para mniej więcej w tym samym stopniu co w meta. Grupy NH2Me+, NHMe2+ i NMe3+ dają więcej pochodnych meta niż para, a udział produktu para podstawionego zmniejsza się wraz z rosnąca liczbą grup metylowych.
- Grupy nie posiadające pary elektronowej na atomie połączonym z pierścieniem i które kierują podstawienie w pozycje orto i para. W tej kategorii znajdują się grupy alkilowe, arylowe i grupa COO–, czyli podstawniki aktywujące pierścień. Grupy arylowe są grupami aktywującymi i kierującymi w pozycje orto i para. Można to wyjaśnić tym, że para elektronów z aromatycznego sekstetu odgrywa rolę wolnej pary elektronowej generując końcowe formy rezonansowe na powyższym schemacie. Wpływ ujemnie naładowanych grup, takich jak COO– jest związany z ich zdolnością do oddawania elektronów, ponieważ nie ma interakcji rezonansowej między grupą a pierścieniem. Wpływ grup alkilowych można wyjaśnić w ten sam sposób, ale dodatkowo możemy również narysować formy kanoniczne wynikające z hiperkoniugacji. Ten efekt skutkuje aktywacją i kierowaniem podstawników w pozycje orto i para. Innym sposobem spojrzenia na wpływ grup alkilowych jest to, że jony orto i para arenu są bardziej stabilne, ponieważ każda z nich zawiera formę, która jest trzeciorzędowym karbokationem, podczas gdy wszystkie formy kanoniczne dla jonu meta są karbokationami drugorzędowymi.
Trudno jest jednoznacznie określić, który z produktów orto czy para będzie przeważał w reakcji substytucji, proporcje tych izomerów zależą od warunków prowadzenia reakcji. Z jednej strony pozycje orto w pierścieniu benzenowym są dwie a para tylko jedna. Z drugiej strony efekt stabilizujący w pozycji para jest większy niż w orto. Biorąc to pod uwagę możemy oczekiwać, że statystycznie udział para podstawionego produktu będzie większy niż 33%, a orto mniejszy niż 67%. Istotnym czynnikiem są również efekty steryczne. W przypadku gdy grupa elektrofilowa oraz podstawnik w pierścieniu są duże to powstawanie produktu orto podstawionego jest utrudnione. Podobna sytuacja występuje gdy podstawnikiem kierujący w pozycje orto i para jest grupa posiadająca pary elektronowe. W tym wypadku również udział związku para podstawionego jest większy niż orto. W przypadku gdy w pierścieniu benzenowym mamy więcej niż jeden podstawnik zachodzi wzajemne wzmocnienie ich działania.
- Silnie aktywujący podstawnik konkuruje ze słabszą lub dezaktywującą grupą narzucając kierunek podstawienia; o-krezol daje podstawienie głównie w pozycji orto i para względem grupy hydroksylowej, a nie metylowej. Możemy uporządkować podstawniki w następującej kolejności: NH2, OH, NR2, O– > OR, OCOR, NHCOR > R, Ar > halogen> grupy kierujące w położenie meta.
- Jeśli obydwa podstawniki są takie same to jest mało prawdopodobne, że trzeci podsatwnik zajmie pozycję meta w stosunku do już obecnych w pierścieniu. Wynika to z zawady przestrzennej, a prawdopodobieństwo maleje wraz z wielkością podstawników znajdujących się w pierścieniu i wielkością atakującego nukleofila.
- Kiedy podstawnik kierujący w pozycję meta zajmuje położenie meta w stosunku do grupy kierującej w pozycje orto i para, kolejny podstawnik zajmuje położenie orto w stosunku do grupy kierującej w pozycję meta (efekt orto).
Substytucja elektrofilowa związków alifatycznych
W przypadku układów aromatycznych najpowszechniejszą grupą opuszczającą jest proton. Proton jest również grupą opuszczającą w układach alifatycznych, ale ich reaktywność zależy od kwasowości. Protony w nasyconych alkanach są niereaktywne, ale podstawienie elektrofilowe często zachodzi przy kwaśnych protonach, na przykład protonach a w stosunku do grupy karbonylowej lub w pozycji alkinylowej (RC≡CH). Ponieważ jony metali są w stanie łatwo przenosić ładunki dodatnie, można się spodziewać, że związki metaloorganiczne będą szczególnie podatne na podstawienie elektrofilowe.
Dwucząsteczkowe mechanizmy elektrofilowej substytucji alifatycznej są analogiczne do mechanizmu SN2, ponieważ nowe wiązanie tworzy się w miarę zrywania starego. W mechanizmie SN2 nadchodząca grupa niesie ze sobą parę elektronów, a orbital na którym są one zlokalizowane może nakładać się orbital węgla tylko w takim stopniu, w jakim grupa opuszczająca zabiera ze sobą elektrony. W innym przypadku węgiel miałby więcej niż osiem elektronów na swojej zewnętrznej powłoce. Ponieważ chmury elektronowe wzajemnie się odpychają więc nadchodząca grupa zbliża się od drugiej strony grupy opuszczającej, powodując odwrócenie konfiguracji. Natomiast gdy nukleofil przekazuje elektrony elektrofilowi przewidzenie kierunku ataku nie jest tak oczywiste. Można założyć dwie możliwości: zbliżanie się elektrofila od przodu SE2 (przód), oraz zbliżanie się elektrofila od strony przeciwnej do grupy opuszczającej SE2 (tył). Można to przedstawić schematycznie w sposób następujący:
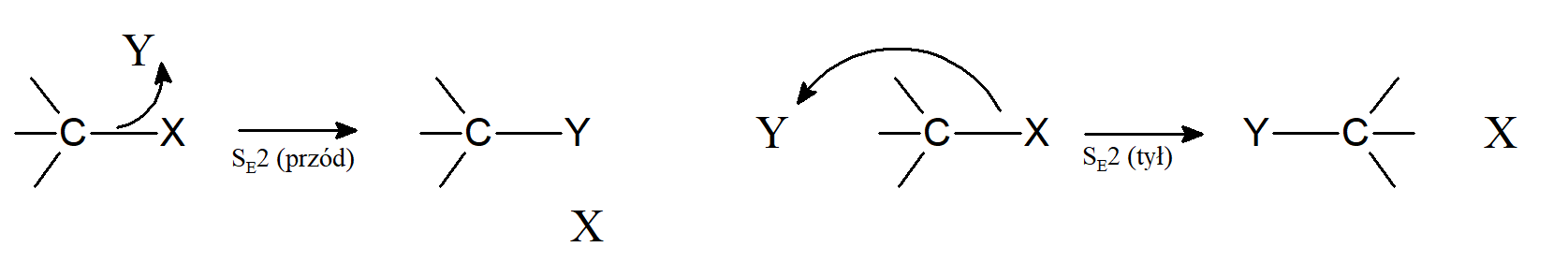
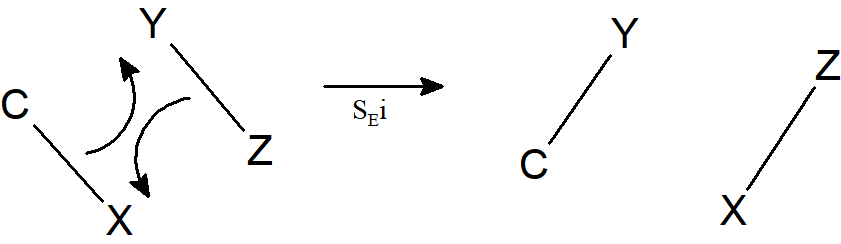
W przypadku gdy elektrofil reaguje zgodnie z mechanizmem SE1 (przód) istnieje trzecia możliwość. Część elektrofilu może uczestniczyć w usunięciu grupy opuszczającej, tworząc z nią wiązanie w tym samym czasie, gdy tworzy się nowe wiązanie C–Y; tego typu mechanizm jest oznaczany jako SEi.
Mechanizm jednocząsteczkowej substytucji elektrofilowej jest analogiczny do mechanizmu SN1 i obejmuje dwa etapu:
R–X → R– + X+ (powoli)
R– + Y+ → R–Y
Substytucja elektrofilowa połączona z migracją wiązania podwójnego
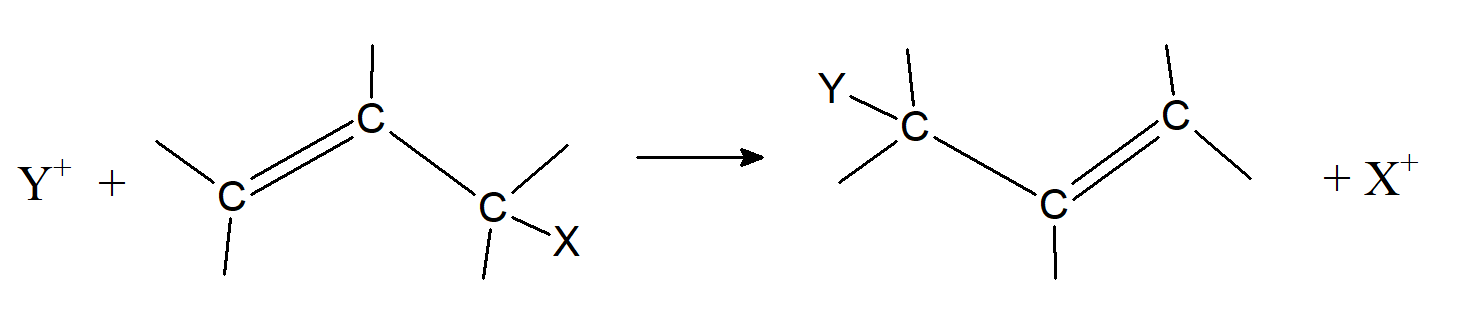
W reakcji podstawienia elektrofilowego związków allilowych może prowadzić do migracji wiązania podwójnego. Proces może przebiegać według dwóch mechanizmów. Pierwszy z nich jest analogiczny do mechanizmu SE1, w którym w pierwszym etapie uwalniana jest grupa opuszczająca tworząc stabilizowany rezonansowo allilowy karboanion, który następnie jest atakowany przez elektrofil.
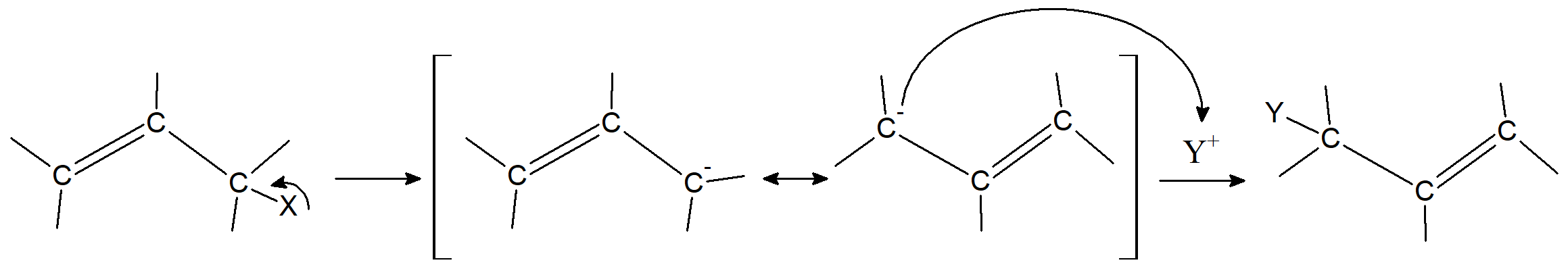
Drugi mechanizm polega na ataku grupa Y przez wiązanie π, dając karbokation, który następnie traci X wraz z utworzeniem jednostki alkenowej.
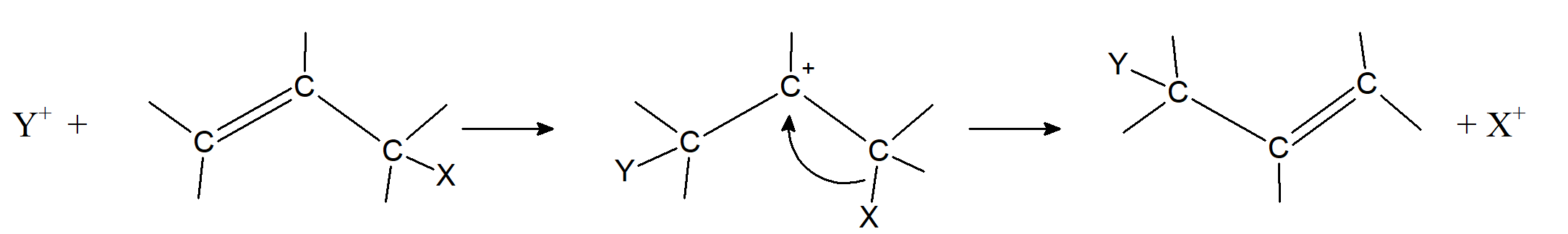
W przypadku reakcji SE1 grupy elektronodonorowe zmniejszają, a grupy elektronoakceptorowe zwiększają szybkość reakcji. Zarówno w przypadku mechanizmu SE1 jak i SE2 im bardziej polarne jest wiązanie C–X, tym łatwiej zachodzi odszczepienie grupy opuszczającej.