Principles of Chemistry
Principles of Chemistry
 Substytucja związków heteroaromatycznych
Substytucja związków heteroaromatycznych
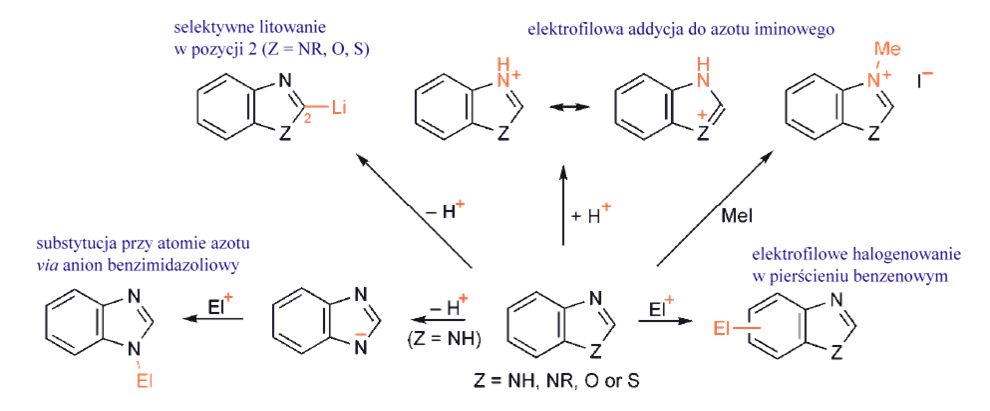
Elektrofilowa substytucja do azotu
W niektórych, zwłaszcza pięcioczłonowych heterocyklach, azot może posiadać przyłączony wodór. istotne jest zrozumienie chemii takich heterocykli zawierających azot, aby zdać sobie sprawę, czy i w jakim stopniu są one zasadowe – będą tworzyć sole z kwasami protonowymi lub kompleksy z kwasami Lewisa – a w przypadku heterocykli z N–H, w jakim stopniu tracą wodór przy azocie w postaci protonu tworząc odpowiednio mocną zasadę. Jako miarę tych właściwości stosujemy wartości pKa do wyrażenia kwasowości heterocykli z NH oraz wartości pKaH do wyrażenia mocy zasady. Im niższa wartość pKa, tym bardziej kwaśny; im wyższa wartość pKaH, tym bardziej zasadowy.
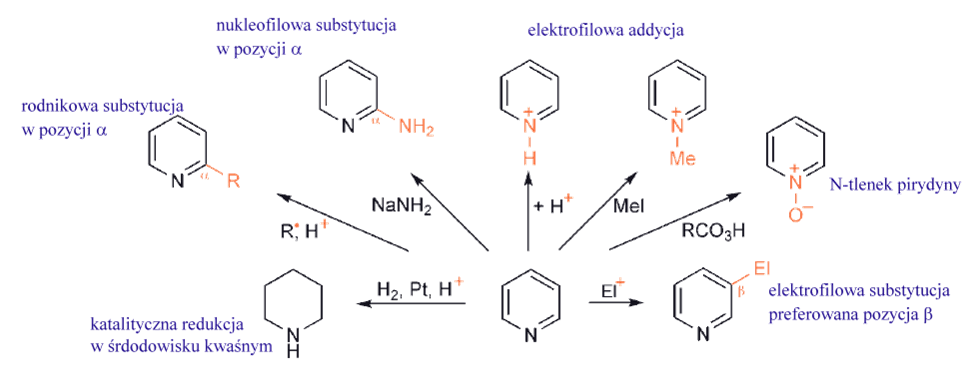
Heterocykle, które zawierają jednostkę iminową (C=N) jako część swojego pierścienia, pirydyny, chinoliny, izochinoliny, 1,2- i 1,3-azole, itp., nie uwzględniają wolnej pary elektronowej azotu w aromatycznym układzie π-i dlatego jest ona dostępna elektrofilom, tak jak w przypadku każdej prostszej aminy. Innymi słowy, takie heterocykle są zasadowe i będą reagować z protonami lub innymi indywiduami elektrofilowymi przy azocie.W wielu przypadkach produkty z takich adduktów – sole – są izolowane.
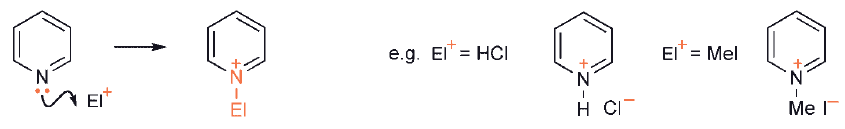
W przypadku addycji protonu równowaga zależy od pKaH heterocyklu, a na to z kolei wpływają podstawniki obecne w pierścieniu: grupy elektronodonorowe zwiększają zasadowość a podstawniki elektronoakceptorowe zmniejszą zasadowość. pKaH prostych pirydyn jest rzędu 5, podczas gdy dla 1,2- i 1,3-azoli zależy od charakteru drugiego heteroatomu: pirazol i imidazol, z dwoma atomami azotu, mają wartości 2,5 i 7.1. Powiązana z zasadowością, ale z pewnością nie zawsze ją odzwierciedlająca, jest N-nukleofilowość heterocykli zawierających ugrupowanie iminowe. Tutaj obecność podstawników sąsiadujących z azotem może mieć znaczny wpływ na to, jak łatwo zachodzi reakcja np. z halogenkami alkilowymi, a nawet czy azot jest zdolny do tworzenia soli.
Podstawienie elektrofilowe cząsteczek aromatycznych, i heteroaromatycznych, następuje według mchanizmu dwuetapowego. W początkowej addycji uczestniczy dodatnio naładowany związek pośredni (kompleks σ- lub związek pośredni Whelanda), a w kolejnym etapie mamy eliminację (zwykle H+). Pierwszy etap jest zwykle wolniejszy. W większości przypadków takie podstawienia są nieodwracalne, a ilość produktu jest określona kinetyką reakcji.
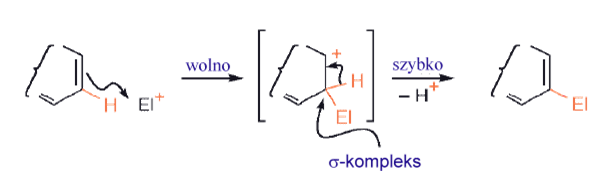
Rozważając heteroaromatyczną substytucję elektrofilową, należy dokonać początkowo podziału na te heterocykle, które są zasadowe i te, które takimi nie są. W pierwszym przypadku, oddziaływaniem wolnej pary elektronowej azotu z elektrofilem, a nawet z jakimkolwiek innym czynnikiem elektrofilowym w mieszaninie reakcyjnej (protony w mieszaninie nitrującej lub chlorek glinu w reakcji Friedla-Craftsa) zachodzą znacznie szybciej niż jakiekolwiek podstawienie przy atomie węgla, przekształcając w ten sposób substrat w dodatnio naładowaną sól, a zatem redukując jego podatność na atak elektrofila na węgiel. Wszystkie azotowe heterocykle typu pirydyny (tj. zawierające C=N) nie ulegają łatwo C-podstawieniu elektrofilowemu, chyba że: (i) w pierścieniu znajdują się inne podstawniki, które go „aktywują” lub (ii) cząsteczka ma inny, skondensowany pierścień benzenowy, w którym może nastąpić substytucja. Na przykład proste pirydyny nie podlegają wielu podstawieniom elektrofilowym, ale chinoliny i izochinoliny ulegają podstawieniu w pierścieniu benzenowym. Oszacowano, że reaktywność pirydyny (nieprotonowanej) w procesie podstawienia elektrofilowego jest około 107 razy mniejsza niż reaktywność benzenu.
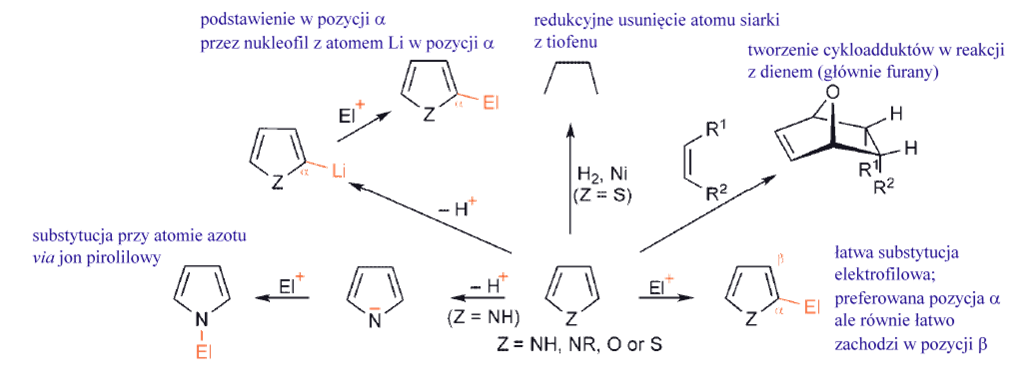
Ppięcioczłonowe, bogatych w elektrony heterocykle, znacznie łatwiej ulegają podstawieniu elektrofilowemu. Związki, takie jak pirol, tiofen i furan, z łatwością przechodzą szereg podstawień elektrofilowych w obu pozycjach pierścienia, ale z preferencją ataku na węgiel sąsiadujący z heteroatomem, czyli w pozycji α-.
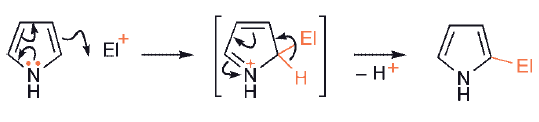
Pirole są bardziej aktywne niż furany, a te z kolei bardziej aktywne niż tiofeny.
Nukleofilowa substytucja przy atomie węgla
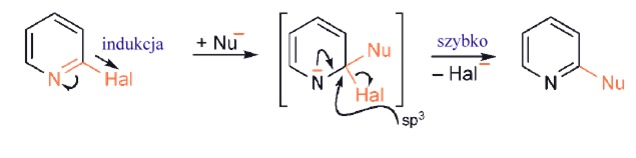
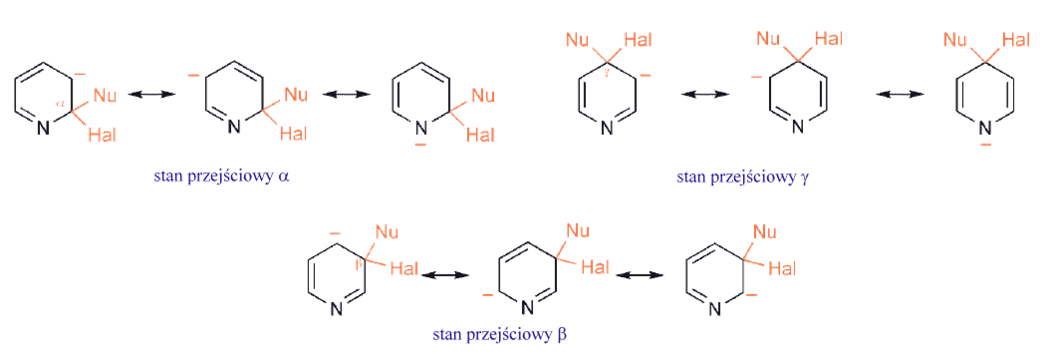
W związkach heterocyklicznych podstawienie grupy opuszczającej, często halogenkowej, przez nukleofil jest istotnym procesem, zwłaszcza dla układów sześcioczłonowych. W chemii pięcioczłonowych aromatycznych heterocykli takie procesy mają znaczenie tylko w sytuacjach, gdy, jak w chemii benzenu, grupa opuszczająca jest aktywowana przez grupę nitrową w pozycji orto- lub para, lub w azolach, gdzie opuszczająca grupa jest przyłączona do węgla jednostki iminowej analogicznie do sześcioczłonowych imin. Pozycje α - i γ - pierścienia sześcioczłonowego halo - azyny są aktywowane w początkowym etapie addycji nukleofilowej przez dwa czynniki: (i) indukcyjny i mezomeryczny związany ze ściaganiem elektronów przez azot oraz (ii) indukcyjne przyciąganie elektronów przez halogen. Dodatkowo w powstałych półproduktach ładunek ujemny znajduje się w dużej mierze na azocie: halogenki α - i γ - są znacznie bardziej reaktywne na podstawienie nukleofilowe niż β–halogenki.
Substytucja rodnikowa na atomie węgla
Reakcja rodników nukleofilowych, w warunkach kwasowych, z heterocyklami zawierającymi jednostkę iminową jest zdecydowanie najważniejszym i najbardziej użytecznym syntetycznie podstawieniem rodnikowym związków heterocyklicznych. Pirydyny, chinoliny, diazyny, imidazole, benzotiazole i puryny należą do układów, dla których wykazano, że reagują z szerokim zakresem rodników nukleofilowych, selektywnie w pozycjach α i γ względem azotu, z wymianą wodoru. Warunki kwasowe są niezbędne, ponieważ N-protonowanie heterocyklu zarówno znacznie zwiększa jego reaktywność, jak i sprzyja regioselektywności wobec rodnika nukleofilowego.
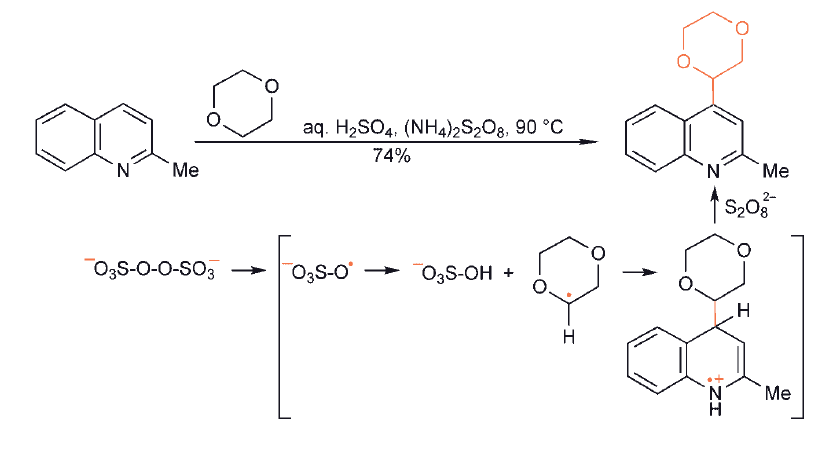
Gdy w substracie heterocyklicznym dostępna jest więcej niż jedna pozycja reaktywna, jak to często ma miejsce w przypadku np. pirydyn, występują potencjalne problemy z regioselektywnością lub/i dissubstytucją (ponieważ produkt pierwszego podstawienia jest często tak samo reaktywny jak związek wyjściowy ). Regioselektywność zależy w pewnym stopniu od charakteru atakującego rodnika i rozpuszczalnika,
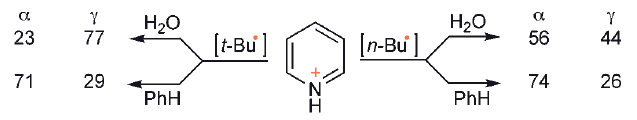
Selektywne monopodstawienie można osiągnąć przez zastosowanie czwartorzędowej soli N+-metoksy, zamiast zwykłej sprotonowanej soli. Tutaj ponowna aromatyzacja jest wynikiem utraty metanolu, pozostawiając jako produkt znacznie mniej reaktywną, obojętną pirydynę.
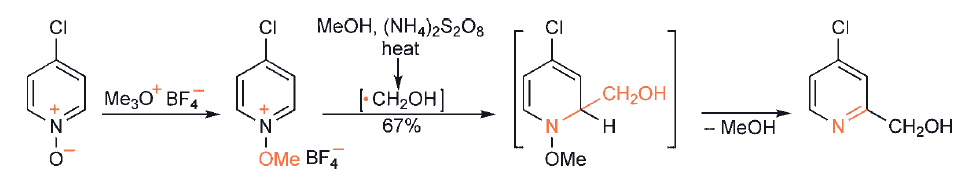
Pirydyny, chinoliny, izochinoliny
Podstawienie elektrofilowe na węglu, przynajmniej w prostych pirydynach, jest bardzo trudne, w przeciwieństwie do reakcji acylowania benzenu; reakcja Friedla – Craftsa np. w przypadku pirydyn w ogóle nie zachodzi. Tę nieaktywność można przypisać dwóm czynnikom:
- Reakcja pirydyny z odczynnikiem zawierającym elektrofile natychmiast przekształca heterocykl w kation pirydyniowy, z elektrofilem (lub protonem z roztworu) przyłączonym do azotu. Stopień konwersji zależy od charakteru i stężenia elektrofila (lub protonów) oraz zasadowości konkretnej pirydyny i jest zwykle prawie całkowity. Oczywiście dodatnio naładowany kation pirydyniowy jest o wiele bardziej odporny na atak elektrofila niż pierwotny obojętny heterocykl. Elektrofil zatem musi albo zaatakować już naładowany dodatnio związek, albo odszukać obojętną pirydynę o bardzo niskim stężeniu.
- Węgle w pierścieniu pirydyny są ubogie w elektrony, szczególnie w pozycjach α i γ: tworzenie kompleksu σ między pirydyną a elektrofilem jest z natury niekorzystne. Najmniej niekorzystną opcją jest atak w pozycji β.
Silnie elektronoakceptorowe podstawniki sprawiają, że pirydyna staje się bardziej obojętna, jednak grupy aktywujące – aminowa i oksy, a nawet alkilowa – pozwalają na substytucję za pośrednictwem protonowanego heterocyklu, tj. poprzez dwukationowy związek pośredni. Obecność podstawników halogenowych, które działają osłabiająco na zasadę i tylko słabo dezaktywują, może umożliwić podstawienie w inny sposób – poprzez zwiększenie stężenia wolnej, obojętnej pirydyny. Pierścienie pirydynowe są odporne na utleniające otwarcie, podobnie jak pierścienie benzenowe. Jednak układ heterocykliczny jest łatwo redukowany katalitycznie, zwłaszcza w roztworze kwaśnym. Podobnie sole pirydyniowe można łatwo redukować zarówno wodorem na katalizatorze, jak i reduktorami nukleofilowymi. Obnizona gęstość elektronowa na węglach w pirydynach, szczególnie w pozycjach α- i γ-, sprawia, że addycja nukleofilowa, a zwłaszcza nukleofilowe podstawienie halogenku (i innych dobrych grup opuszczających), jest bardzo ważną cechą chemii pirydyn.
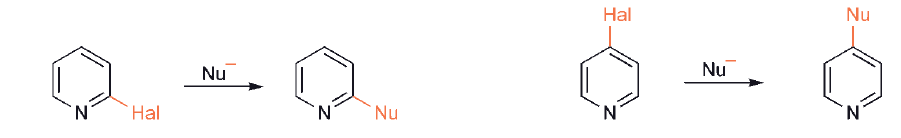
Addycja jest ułatwiona przez: (i) niedobór elektronów przy węglach α i γ, dodatkowo zwiększony przez podstawnik halogenowy, oraz (ii) zdolność heteroatomu do przyjmowania ładunku ujemnego co wpływa na szybkość powstawania półproduktu. Porównanie trzech możliwych związków pośrednich od razu pokazuje, że ten ostatni nie jest dostępny do ataku w pozycji β, a zatem substytucja nukleofilowa w pozycji β jest znacznie wolniejsza i praktycznie nie występuje.
W przypadku braku halogenu w pozycjach α- lub γ-, pirydyny są mniej reaktywne i, oczywiście, nie mają podstawnika odpowiedniego do uwolnienia w formie anionu w celu zakończenia podstawienia nukleofilowego. Występują jednak addycje nukleofilowe, ale powstały addukt dihydropirydynowy wymaga w pewien sposób usunięcia „wodorku”, w celu zakończenia podstawienia. Takie reakcje, na przykład z amidkiem sodu lub z reagentami metaloorganicznymi, są selektywne dla pozycji α, prawdopodobnie dlatego, że nukleofil jest dostarczany przez kompleks obejmujący oddziaływanie azotu pierścienia z kationem metalu związanym z nukleofilem.
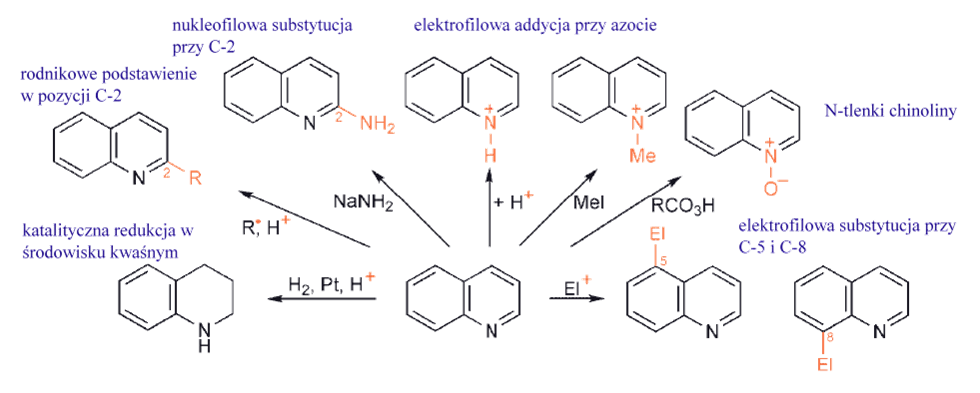
Chinolina i izochinolina reagują z elektrofilami w obrębie pierścieni benzenowych; z nukleofilami reakcja przebiega w obrebie pierścienia pirydynowego, zwłaszcza w pozycjach α i γ względem azotu, i są bardziej reaktywne niż pirydyny.
Diazyny
Pirydazyna, pirymidyna i pirazyna zawierają dwa iminowe atomy azotu. Dwa heteroatomy ściągają gęstość elektronową z węgli pierścienia w większym stopniu niż jeden atom N w pirydynie, więc niepodstawione diazyny są bardziej odporne na podstawienie elektrofilowe niż pirydyna. Konsekwencją jest to, że ten sam zwiększony niedobór elektronów na węglu sprawia, że diazyny są łatwiej atakowane przez nukleofile niż pirydyna. Dostępność wolnych par elektronowych azotu jest również zmniejszona: każda z diazyn jest znacznie mniej zasadowa niż pirydyna, co odzwierciedla destabilizujący wpływ drugiego azotu. Niemniej jednak diazyny będą tworzyć sole i będą reagować z halogenkami alkilu oraz nadkwasami, dając odpowiednio N-alkilowe czwartorzędowe sole i N-tlenki. Mówiąc ogólnie, takie addycje elektrofilowe mają miejsce tylko przy jednym azocie, ponieważ obecność ładunku dodatniego w produktach powoduje, że drugi azot jest wyjątkowo niereaktywny w stosunku do drugiego addycji elektrofilowej.

Pirol, furan, tiofen
W chemii pirolu i tiofenu dominuje substytucja elektrofilowa, preferencyjnie w pozycji α, oraz w pozycji β, w przypadku gdy pozycja α jest zajęta przez podstawnik.
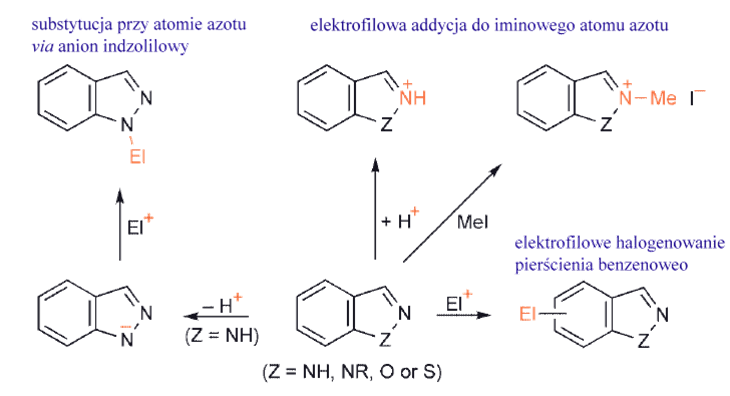
1,2-, 1,3-azole oraz benzoazole
1,2- są mniej nukleofilowe i mniej zasadowe przy azocie iminowym niż ich 1,3-izomery. To, że elektrofilowe addycje występują łatwo pokazuje, że wolna para elektronowa na azocie iminowym nie jest zaangażowana w aromatyczny sekstet elektronowy.
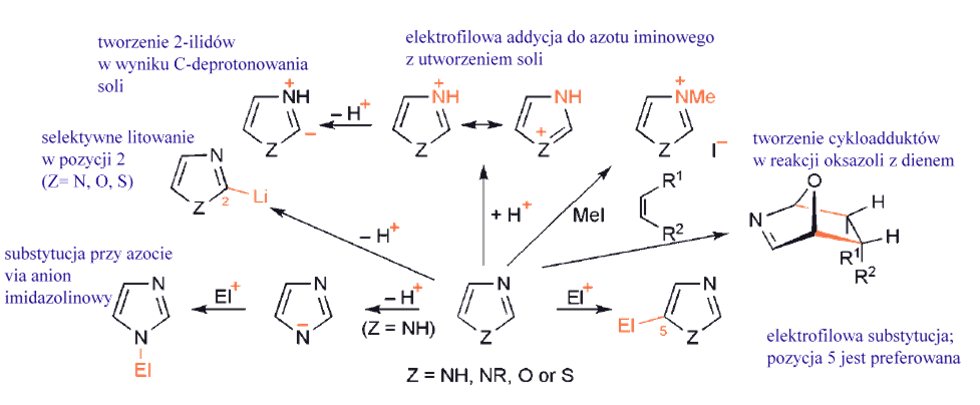
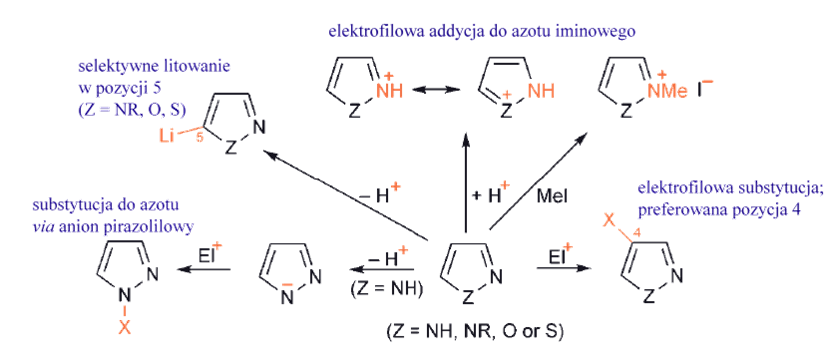
Benzo-1,3- i -1,2-azole nie ulegają podstawieniu elektrofilowemu w heteropierścieniu; Te bicykliczne związki są nieco słabszymi zasadami niż azole, ale łatwo reagują z halogenkami alkilowymi dając sole.