Principles of Chemistry
Principles of Chemistry
 Reakcje substytucji
Reakcje substytucji
Substytucja alifatyczna
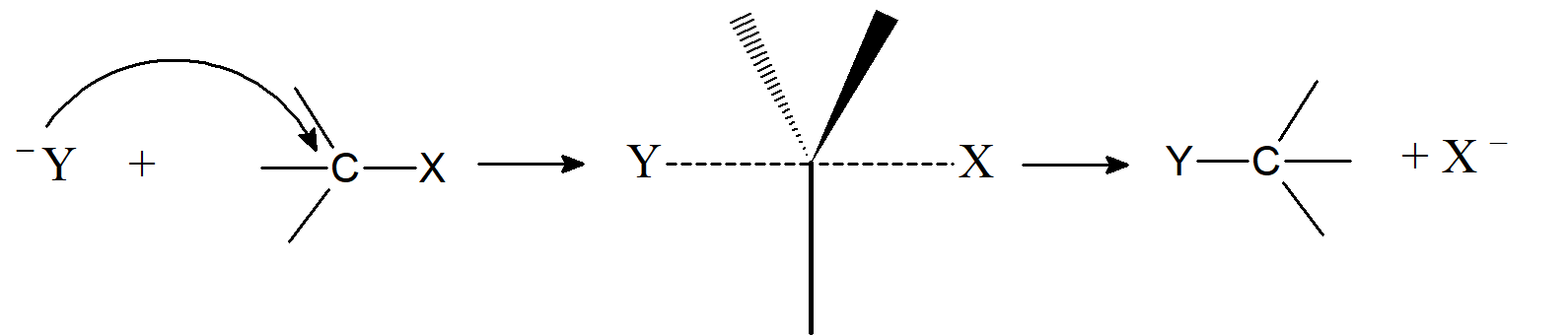
W nukleofilowej substytucji alifatycznej atakujący (oddający elektrony) odczynnik (nukleofil) przenosi parę elektronów do substratu, używając tej pary do utworzenia nowego wiązania, a grupa opuszczająca odchodzi z parą elektronów:
![]()
Przedstawione równanie nic nie mówi o ładunkach. Nukleofil Y może być obojętny lub naładowany ujemnie; RX może być obojętne lub naładowane dodatnio; więc istnieją cztery typy tych reakcji, których przykładami są:
R–I + OH– → R–OH + I–
R–I + NMe3 → R–+NMe3 + I–
R–+NMe3 + OH– → R–OH + NMe3
R–+NMe3 + H2S → R–+SH2 + NMe3
We wszystkich przypadkach Y musi mieć wolną parę elektronową, gdyż wszystkie nukleofile są zasadami Lewisa. Gdy Y jest rozpuszczalnikiem, reakcję nazywa się solwolizą. Podstawienie nukleofilowe na węglu alkilowym nosi nazwę alkilowania nukleofilu; podstawienie nukleofilowe na węglu acylowym jest acylowaniem nukleofila.
Najczęściej proces przebiega według mechanizmu SN2 czyli jest to reakcja dwucząsteczkowa. Reakcja rozpoczyna się od ataku nukleofila na atom centralny z przeciwnej strony, niż zajmuje grupa odchodząca. W przypadku reakcji przy atomie węgla powstaje stan przejściowy o budowie podwójnej piramidy trygonalnej, w której atom ten jest chwilowo pięciowiązalny, a trzy podstawniki, które nie biorą udziału w reakcji, umiejscowione są w jednej płaszczyźnie, prostopadłej do osi wiązań nukleofil – atom węgla – grupa odchodząca. W drugim etapie od stanu przejściowego odrywa się grupa odchodząca, atom węgla staje się ponownie czterowiązalny, a trzy podstawniki, nie biorące udziału w reakcji, ulegają przemieszczeniu w stronę grupy odchodzącej.
Drugim rodzajem jest
substytucja nukleofilowa zachodząca poprzez mechanizm jednocząsteczkowy SN1. Reakcje te polegają na wymianie atomu lub grupy atomów na inne pod wpływem działania nukleofila, przy czym etapem kluczowym (decydującym o szybkości i kierunku procesu) jest oderwanie od centralnego atomu grupy opuszczającej z utworzeniem mniej lub bardziej trwałego kationu
R–X ↔ R+ + X
który następnie szybko reaguje z nukleofilem
R+ + Y → R–Y
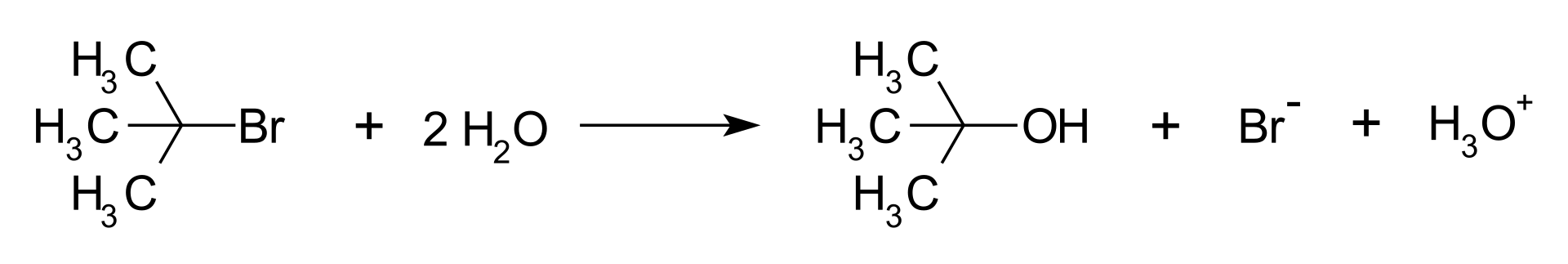
Przykładem reakcji SN1 jest hydroliza bromku tert-butylowego w wyniku której powstaje alkohol tert-butylowy, anion bromkowy i jon hydroniowy.
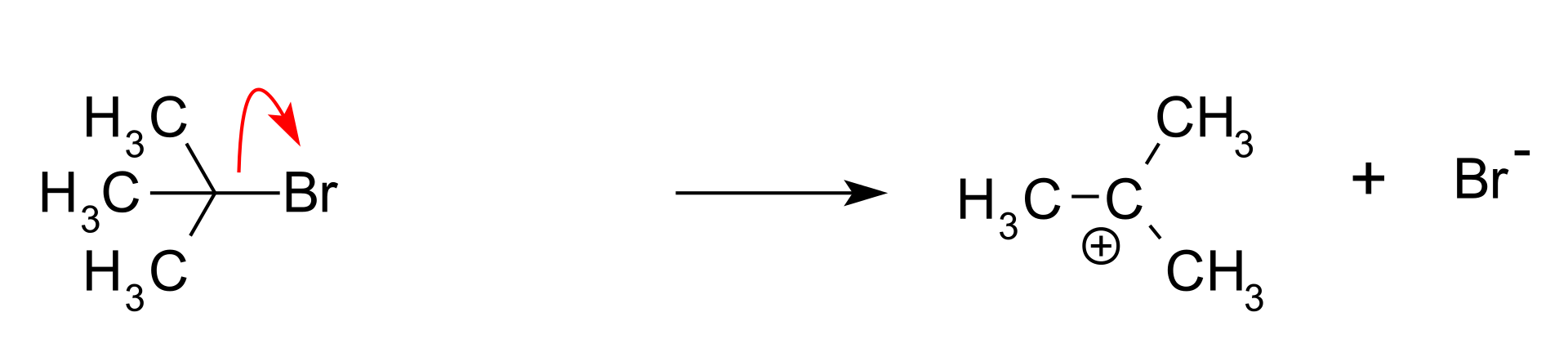
Reakcja ta zachodzi w trzech etapach:
– Tworzenie karbokationu:
– Atak nukleofila: Karbokation reaguje z nukleofilem, w tym przypadku z cząsteczką wody. Nukleofil może zaatakować karbokation z obu stron z równym prawdopodobieństwem, co jest przyczyną racemizacji w przypadku reakcji SN1 związków chiralnych. Jeżeli nukleofil jest anionem, na tym kończy się reakcja, jeśli nie jest, powstaje kolejny produkt przejściowy, w tym przypadku kation t-butylohydroksoniowy:

– Deprotonacja: Reakcję w takim przypadku kończy trzeci etap zwany deprotonacją, polegający na oderwaniu się protonu i powstaniu alkoholu oraz jonu hydroniowego
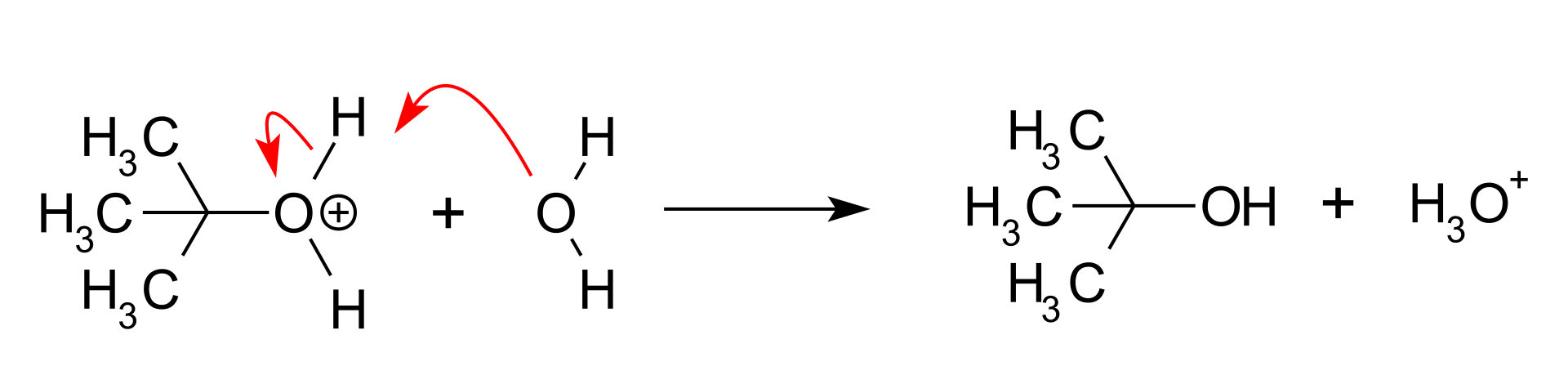
Wszystkie etapy reakcji SN1 są odwracalne. W obecności wody równowaga przesunięta jest w kierunku alkoholu, natomiast w środowisku bezwodnym alkohol tert-butylowy reaguje z bromowodorem z wytworzeniem bromku tert-butylowego.
Mechanizm SET (single electron transfer)
W niektórych reakcjach, w których podstawienie nukleofilowe wydaje się być oczywiste, istnieją dowody, że w rzeczywistości zaangażowane są rodniki i / lub jonorodniki. Pierwszym etapem w takim procesie jest przeniesienie elektronu z nukleofila do substratu z utworzeniem anionorodnika:
R–X + :Y– → R–X·– + Y·
Jonorodnik ulega rozpadowi:
R–X·– → R· + :X–
Powstałe w ten sposób rodniki mogą utworzyć produkt końcowy w reakcji z Y· wytworzonym w pierwszym etapie lub z nukleofilowym jonem Y–, w którym to przypadku konieczny jest dodatkowy etap:
R· + Y· → R–Y
lub
R· + :Y–→ R–Y·–
R–Y·– + R–X → R–Y + R–X·–
W drugim przypadku jon R–X·– powstaje w etapie czwartym jak i pierwszym co pokazuje, że może zajść reakcja łańcuchowa.
Wpływ grup sąsiadujących
W przypadku niektórych substratów szybkość reakcji jest większa niż oczekiwano a konfiguracja na chiralnym atomie węgla zostaje zachowana i nie obserwuje się racemizacji. W takich przypadkach zwykle istnieje grupa posiadająca wolną parę elektronową w pozycji b w stosunku do grupy opuszczającej. Mechanizm działający w takich przypadkach jest determinowany obecnością grup sąsiadujących i zasadniczo składa się z dwóch kolejnych podstawień typu SN2, z których każda powoduje inwersję, więc wynikiem końcowym jest zachowanie konfiguracji. W pierwszym etapie tej reakcji grupa sąsiadująca działa jak nukleofil osłabiając wiązanie grupy opuszczającej. W drugim etapie zewnętrzny nukleofil zajmuje miejsce opuszczone przez grupę opuszczającą.
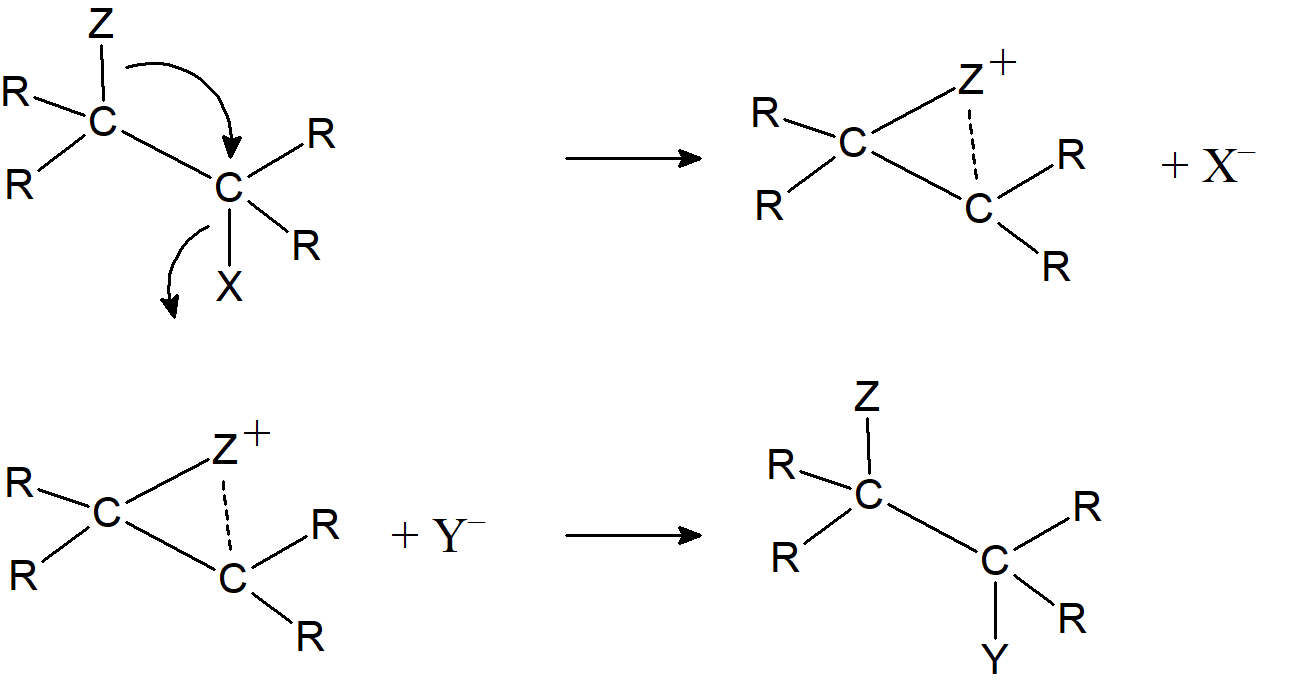
Powodem, dla którego atak Z przebiega szybciej niż atak Y jest to, że grupa Z jest bardziej dostępna. Aby nastąpiła reakcja z Y musi on zderzyć się z cząsteczką substratu, natomiast Z jest łatwo dostępny ze względu na położenie w strukturze cząsteczki. Reakcja między substratem a Y wiąże się z dużym spadkiem entropii aktywacji natomiast reakcja z Z pociąga za sobą znacznie mniejszą utratę entropii.
Do ważniejszych grup sąsiadujących należą COO– (ale nie COOH), COOR, COAr, OCOR, OR, OH, O–, NH2, NHR, NR2, NHCOR, SH, SR, S–, SO2Ph, I, Br i Cl. Skuteczność halogenów jako grup sąsiadujących spada w kolejności I > Br > Cl. Chlor wykazuje bardzo słaby wpływ jako grupa sąsiadująca i wywołuje takie działanie tylko wtedy, gdy zostaje zmarginalizowany wpływ rozpuszczalnika. Na przykład w solwolizie tosylanu 5-chloro-2-heksylu w kwasie octowym, wpływ Cl jest niewielki, ale w znacznie mniej nukleofilowym kwasie trifluorooctowym wpływ Cl jako grupy sąsiedniej staje się decydujący. Grupami sąsiadującymi są tez wiązania podwójne, grupa metylowa oraz atom H.
Reakcje substytucji nukleofilowej w przypadku związków allilowych przebiegają bardzo szybko ale zwykle towarzyszy im przegrupowanie allilowe. Substraty allilowe w reakcji przebiegającej według mechanizmu SN1 zwykle dają dwa produkty: normalny i przegrupowany.
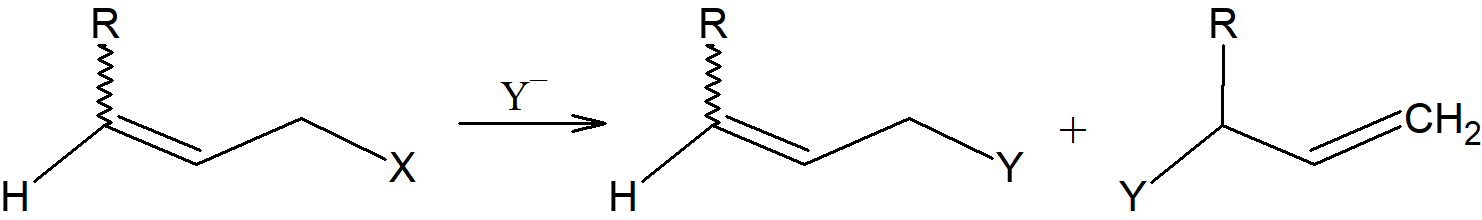
Powstawanie dwóch produktów wynika ze struktury rezonansowej karbokationu R–CH=CH–+CH2 oraz R–+CH-CH=CH2 w wyniku czego węgle C1 i C3 mają ładunek dodatni i mogą być atakowane przez nukleofil Y–. Mechanizm ten określany jest symbolem SN1’.
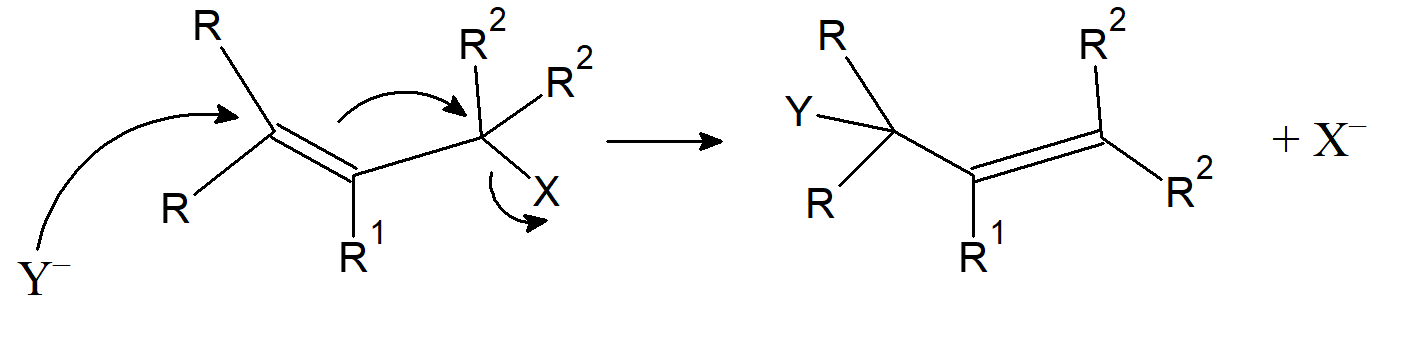
Podstawienie nukleofilowe na atomie węgla allilowego może również zachodzić zgodnie z mechanizmem SN2, w którym to przypadku zwykle nie występuje przegrupowanie allilowe. Jednak przegrupowanie allilowe może również zachodzić w warunkach SN2, poprzez następujący mechanizm, w którym nukleofil atakuje węgiel γ zamiast oczekiwanego węgla z ładunkiem dodatnim:
Podstawienie nukleofilowe na winylowym węglu jest trudne, ale znanych jest wiele przykładów. Najpowszechniejszymi mechanizmami są mechanizm tetraedryczny i mechanizm addycji-eliminacji. Oba te mechanizmy są niemożliwe dla związków nasyconych. Mechanizm tetraedryczny, często nazywany również eliminacją addycyjną (AdN-E), zachodzi znacznie trudniej niż w przypadku związków karbonylowych, ponieważ ujemny ładunek związku pośredniego musi być zlokalizowany na węglu, który jest mniej elektroujemny niż tlen, siarka, lub azot:
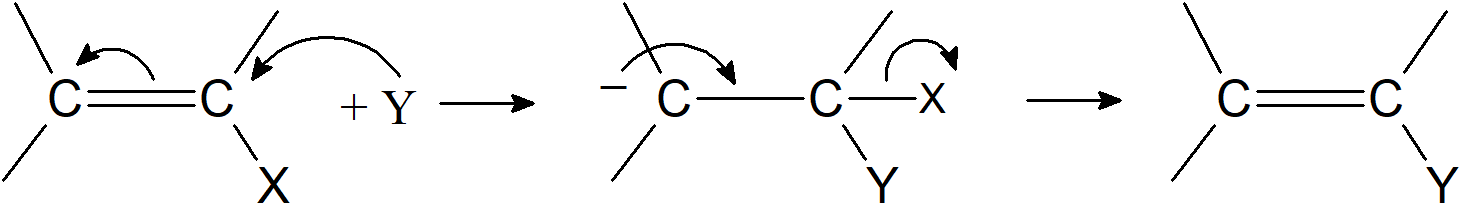
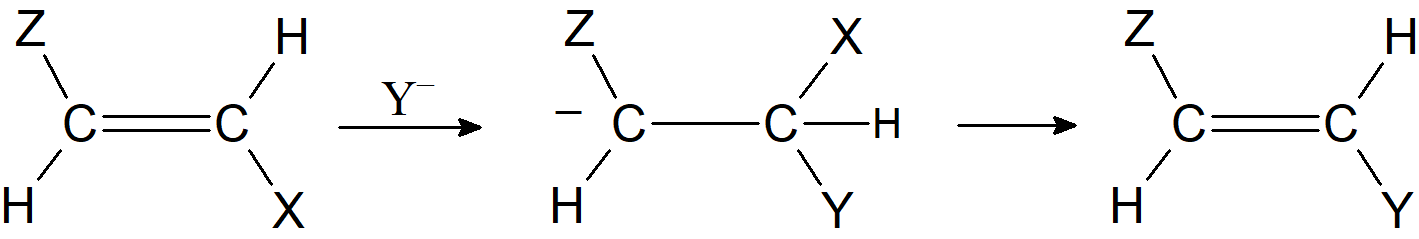
Związek pośredni może zostać ustabilizowany przez połączenie z kationem. Kiedy tak się dzieje, reakcja jest nukleofilową addycją do podwójnego wiązania C=C. Nie jest zaskakujące, że z substratami winylowymi często konkurują addycja i substytucja. Niepodstawione związki winylowe reagują bardzo słabo, jeśli w ogóle, przez te mechanizmy, ale w przypadku gdy mamy do czynienia z układem typu ZCH=CHX, gdzie Z jest grupą odciągającą elektrony, taką jak HCO, RCO, EtOOC, ArSO2, NC i F, reakcje substytucji zachodzą znacznie łatwiej ponieważ grupy przy atomie węgla β stabilizują karboanion:
Wpływ struktury substratu na reaktywność
Wpływ struktury substratu na reaktywność w reakcji nukleofilowej zależy od mechanizmu zgodnie z którym przebiega reakcja. W przypadku mechanizmu SN2 rozgałęzienie na węglu a lub b zmniejsza szybkość reakcji. Związki trzeciorzędowe rzadko reagują poprzez mechanizm SN2, a związki neopentylowe reagują tak wolno, że takie reakcje są generalnie syntetycznie bezużyteczne. Mechanizm tetraedryczny w przypadku związków mających podstawniki przy węglach α lub β w stosunku do węgla karbonylowego również zachodzi znacznie wolniej lub nie zachodzi wcale.
Natomiast w przypadku mechanizmu SN1 obecność podstawników rozgałęziających łańcuch węglowy skutkują zwiększeniem szybkości reakcji; reaktywność kationów maleje w szeregu trzeciorzędowy > drugorzędowy > pierwszorzędowy. Pierwszo– i drugorzędowe związki na ogół reagują zgodnie z mechanizmem SN2, a trzeciorzędowe mechanizmem SN1. Jednak związki trzeciorzędowe rzadko w ogóle ulegają substytucji nukleofilowej. Eliminacja jest zawsze możliwą reakcją uboczną substytucji nukleofilowych (wszędzie tam, gdzie obecny jest wodór b), a w przypadku substratów trzeciorzędowych zwykle przeważa. Z kilkoma wyjątkami podstawienia nukleofilowe na trzeciorzędowym węglu mają niewielką lub żadną wartość preparatywną. Jednak substraty trzeciorzędowe, które mogą reagować w mechanizmie SET (np. p-NO2C6H4CMe2Cl) dają bardzo dobrą wydajność produktów substytucji w reakcjach z różnymi nukleofilami.
Związki winylowe, acetylenowe i arylowe są niereaktywne w stosunku do reakcji substytucji nukleofilowj. W przypadku tych związków oba mechanizmy SN1 i SN2 przebiegają bardzo wolno. Jednym z powodów takiego stanu rzeczy jest to, że atomy węgla sp2 (a nawet sp) mają wyższą elektroujemność niż atomy węgla o hybrydyzacji sp3, a tym samym silniej przyciągają elektrony wiązania. Wiązanie sp–H ma wyższą kwasowość niż wiązanie sp3–H, a kwasowość wiązania sp2–H lokuje się pośrodku. To jest rozsądne; węgiel zatrzymuje elektrony, gdy traci się proton, a węgiel sp, który ma największe powinowactwo do elektronów, najłatwiej traci proton. Jednak w przypadku podstawienia nukleofilowego, grupa opuszczająca zabiera ze sobą parę elektronów, więc sytuacja jest odwrotna i to węgiel sp3 najłatwiej traci grupę opuszczającą z parą elektronową. Reakcje SN1 na związków winylowych mogą zachodzić łatwiej dzięki wpływowi podstawników stabilizujących kation, a reakcje przebiegające zgodnie z mechanizmem tetraedrycznym mogą być przyspieszane przez podstawniki przy węglach b, które stabilizują karboanion. Ponadto reakcje na związków winylowych mogą w niektórych przypadkach przebiegać przez sekwencje addycja - eliminacja lub eliminacja – addycja. Związki typu RCOX reagują łatwiej niż RCH2X, ale odbywa się to głównie w mechanizmie tetraedrycznym. Węgiel karbonylowy ma zwiększony ładunek dodatni co znacznie ułatwia atak nukleofila, a trygonalny atom węgla karbonylowego stanowi mniejszą zawadę przestrzenną dla nukleofila niż węgiel tetraedryczny.
Szybkości reakcji według mechanizmu SN1 zwiększają się gdy przy węglu β występuje wiązanie podwójne; związki allilowe i benzylowe reagują szybko. Powodem jest to, że kationy są stabilizowane przez rezonans. Ogólnie rzecz biorąc, szybkości reakcji typu SN1 związków allilowych są zwiększane w obecności dowolnego podstawnika w pozycji 1 lub 3, który może stabilizować karbokation przez rezonans. Wśród tych podstawników istotną rolę odgrywają grupy alkilowe, arylowe i podstawniki halogenowe. Wiązania potrójne w pozycji b (w układach propargilowych) dają mniej więcej taki sam efekt jak wiązania podwójne. Natomiast grupy alkilowe i halogeny w pozycji 1 zmniejszają szybkość reakcji z powodu zawady przestrzennej.
Związki o wzorze ZCH2X, w których Z = RO, RS lub R2N bardzo szybko ulegają reakcjom według mechanizmu SN1, z powodu zwiększonego rezonansu na karbokationie. Grupy te mają wolną parę elektronową na atomie bezpośrednio przyłączonym do dodatniego węgla, który stabilizuje karbokation. Gdy Z w ZCH2X to RCO, HCO, ROCO, NH2CO, NC lub F3C, szybkości reakcji typu SN1 są mniejsze w porównaniu do CH3X, z powodu ich charakteru destabilizującego układ elektronowy. Karbokationy z grupą CO lub CN są silnie destabilizowane z powodu częściowego ładunku dodatniego na sąsiednim węglu. Gdy Z = SOR lub SO2R podstawienie nukleofilowe przebiega znacznie wolniej. Mechanizm SN1 jest spowolniony przez efekt odciągania elektronów grup SOR lub SO2R, a mechanizm SN2 przypuszczalnie przez efekt steryczny.
Badania reakcji podstawienia serii związków p-ZC6H4CH2X, umożliwiają zbadanie wpływu grup Z na szybkość reakcji substytucji. Efekty steryczne Z są zminimalizowane lub wyeliminowane, ponieważ podstawnik ten znajduje się daleko od miejsca reakcji. W przypadku reakcji SN1 charakter elekronoakceptorowy Z zmniejsza, a podstawnik elektronodonorowy zwiększa szybkość reakcji. Wynika to z tego, że podstawnik elktronodonorowy zmniejszają energię stanu przejściowego (i karbokationu) poprzez rozmycie ładunku dodatniego, na przykład:
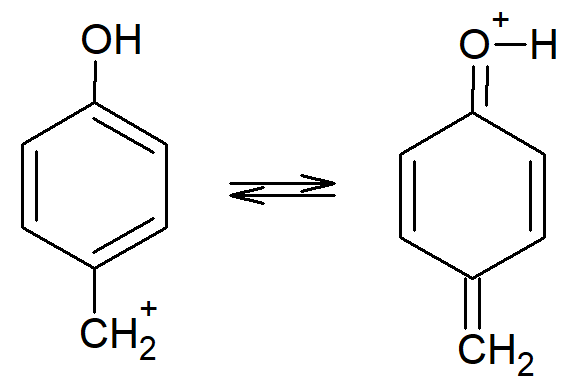
podczas gdy grupy odciągające elektrony koncentrują ładunek na danym fragmencie cząsteczki. W przypadku reakcji SN2 nie znaleziono takich prostych korelacji. Natomiast w przypadku substratów, które reagują zgodnie z mechanizmem tetraedrycznym, grupy elektronoakceptorowe zwiększają szybkość, a grupy elektronodonorowe zmniejszają szybkość reakcji.
Wpływ rodzaju nukleofila na szybkość reakcji substytucji
Każda cząsteczka będąca zasadą Lewisa może być nukleofilem, niezależnie od tego, czy jest obojętna, czy ma ładunek ujemny. Szybkości reakcji SN1 nie zależą od rodzaju nukleofila. Można to zilustrować efektem jaki wywiera zmiana nukleofila z H2O na OH– w reakcji z pierwszorzędowym i trzeciorzędowym substratem. W przypadku bromku metylu, który reaguje zgodnie z mechanizmem SN2, szybkość reakcji z OH– jest większa 5000 razy niż w przypadku reakcji z wodą, ale w przypadku bromku tert-butylu, który reaguje zgodnie z mechanizmem SN1, szybkość reakcji z wodą lub jonem hydroksylowym jest taka sama. Zmiana w nukleofila może jednak prowadzić do zmiany produktu końcowego reakcji SN1. Solwoliza tosylanu benzylu w metanolu daje eter benzylometylowy (nukleofilem jest metanol). Natomiast jeśli zastosuje się silniejszy nukleofil, Br–, szybkość reakcji nie ulega zmianie, ale produktem jest bromek benzylu.
W przypadku reakcji SN2 przebiegających w roztworze istnieją cztery główne zasady rządzące wpływem rodzaju nukleofila na szybkość reakcji, chociaż kolejność nukleofilowości zależy od substratu, rozpuszczalnika, grupy opuszczającej i innych czynników.
– Nukleofil anionowy jest zawsze silniejszym nukleofilem niż sprzężony z nim kwas (zakładając, że ten ostatni jest również nukleofilem). Zatem OH– jest silniejszym nukleofilem niż H2O, NH2 jest silniejszy niż NH3 i tak dalej.
– Porównując nukleofile, których atom donorowy znajduje się w tym samym okresie układu okresowego znajdujemy, że nukleofilowość jest w przybliżeniu zgodna z zasadowością, chociaż zasadowość jest determinowana termodynamicznie, a nukleofilowość jest kinetycznie. Zatem przybliżona kolejność nukleofilowości to NH2– > RO– > OH– > R2NH– > ArO– > NH3 > pirydyna > F– > H2O > ClO4– oraz R3C– > R2N– > RO– > F–.
– Przechodząc w dół grupy, nukleofilowość wzrasta, chociaż zasadowość maleje. Zatem kolejność nukleofilowości halogenków to I– > Br– > Cl– > F–. Podobnie, każdy nukleofil siarkowy jest silniejszy niż jego analog tlenowy, to samo dotyczy fosforu i azotu. Głównym powodem tego rozróżnienia między zasadowością a mocą nukleofilową jest to, że mniejsze ujemnie naładowane nukleofile są silniej solwatowane przez polarne rozpuszczalniki protonowe. Ponieważ ujemny ładunek Cl– jest bardziej skoncentrowany niż ładunek I–, ten pierwszy jest ściślej otoczony powłoką cząsteczek rozpuszczalnika, które stanowią barierę między nim a substratem. Jest to istotne w przypadku protonowych rozpuszczalników polarnych, w których cząsteczki rozpuszczalnika mogą tworzyć wiązania wodorowe ze stosunkowo małymi nukleofilami. Jednak solwatacja nie czynnikiem dającym pełną odpowiedź, ponieważ nawet w przypadku obojętnych nukleofili nukleofilowość wzrasta przechodząc w dół kolumny w układzie okresowym. Leżące niżej w grupach nukleofile nie są tak silnie solwatowane, a zmiany rozpuszczalnika nie wpływają znacząco na ich nukleofilowość. Aby wyjaśnić te przypadki, należy odnieść się do teorii twardych i miękkich kwasów i zasad. Proton jest twardym kwasem, ale substrat alkilowy (który można uważać za kwas Lewisa w stosunku do nukleofila uważanego za zasadę) jest znacznie bardziej miękki, możemy zatem oczekiwać, że grupa alkilowa będzie preferować bardziej miękkie nukleofile niż proton. Zatem większe, bardziej polaryzowalne (bardziej miękkie) nukleofile wykazują większe (względne) powinowactwo do węgla alkilowego niż do protonu.
– Im mniej związany nukleofil, tym większa szybkość reakcji. Szybkość ataku (EtOOC)2CBu– Na+ w benzenie zwiększa się przez wprowadzenie substancji (np. 1,2 -dimetoksyetan, adipamid), które specyficznie solwatują Na+ pozostawiając swobodny anion. W niepolarnym rozpuszczalniku, takim jak benzen, sole, takie jak (EtOOC)2CBu– Na+, występują zwykle w postaci agregatów par jonowych o dużych masach cząsteczkowych.
W przypadku podstawienia na węglu karbonylowym, kolejność nukleofilowości nie jest taka sama, jak w przypadku substytucji na węglu nasyconym, wykazuje większą zgodność z szeregiem zasadowości. Przyczyną jest przypuszczalnie to, że węgiel karbonylowy ze swoim częściowym ładunkiem dodatnim bardziej przypomina proton niż węgiel w związku nasyconym, co oznacza, że węgiel karbonylowy jest znacznie twardszym kwasem niż węgiel z wysyconymi wiązaniami. Kolejność nukleofilowości dla substratów karbonylowych jest następująca: Me2C=NO– > EtO– > MeO– > OH– > ArO– > N3– > F– > H2O> Br– > I–. Miękkie zasady są w niewielkim stopniu atakują węgiel karbonylowy. Obecność niebiorących udziału w tworzeniu wiązań par elektronowych na atomie nukleofilowym zwiększa aktywność nukleofila.
Wpływ grup opuszczających
Grupa odchodząca w reakcji substytucji nukleofilowej ulega odszczepieniu tym łatwiej, im bardziej stabilna jest jako odrębna jednostka. Zwykle łatwość odszczepienia grupy opuszczającej jest to odwrotna do jej zasadowości, czyli najłatwiej ulegają odszczepieniu grupy będące słabymi zasadami. Jodek jest najlepszą grupą opuszczającą spośród halogenków, a odszczepienie fluorku jest najtrudniejsze. Ponieważ XH jest zawsze słabszą zasadą niż X–, podstawienie nukleofilowe jest zawsze łatwiejsze w przypadku substratu RXH+ niż RX. Przykładem jest to, że grupy OH i OR nie są odszczepiane od alkoholi i eterów, ale proces taki następuje po sprotonowaniu tych gup czyli przekształceniu w ROH2+ lub RORH+. Reakcje, w których grupa opuszczająca nie ulega oderwaniu do momentu sprotonowania nazywane są SN1cA lub SN2cA, w zależności od tego, czy po sprotonowaniu reakcja przebiega według mechanizmu SN1 czy SN2 (oznaczenia te są często skracane do A1 i A2). Oczywistym jest, że najsilniejsze nukleofile (np. NH2–, HO–) nie mogą brać udziału w procesach SN1cA lub SN2cA, ponieważ uległyby konwersji do ich sprzężonych kwasów w warunkach kwasowych niezbędnych do protonowania grup opuszczających. Ponieważ reakcje SN1 nie wymagają silnych nukleofilów, ale wymagają dobrych grup opuszczających, większość z nich zachodzi w warunkach kwaśnych. Natomiast reakcje SN2, które wymagają silnych nukleofili (które są na ogół silnymi zasadami), najczęściej zachodzą w środowisku zasadowym lub obojętnym. Chociaż halogenki są powszechnymi grupami opuszczającymi w substytucji nukleofilowej, często wygodniej jest stosować alkohole. Ponieważ grupę OH trudno jest odszczepić od alkoholu należy ją sprotonować lub przekształcić alkohol do reaktywnego estru, najczęściej estru sulfonowego. Podobnie jak grupa hydroksylowa tak grupy NH2, NHR, NR2 są trudne do usunięcia w reakcji substytucji. Również w tych przypadkach przekształca się je w tosylany. Bardzo dobrą grupą opuszczającą jest N2 z układu RN2+.
Wpływ rozpuszczalnika na szybkość reakcji substytucji nukleofilowej
Wpływ polarności rozpuszczalnika na szybkość reakcji SN1 zależy od tego, czy substraty reakcji są obojętne czy kationowe. W przypadku związków obojętnych, które stanowią większość przypadków, im bardziej polarny rozpuszczalnik, tym szybsza reakcja, ponieważ ładunek karbokationu jest większy w stanie przejściowym niż w związku wyjściowym, a energia jonowego stanu przejściowego jest obniżana w rozpuszczalnikach polarnych. Jednak w przypadku substratów kationowych, ładunek w stanie przejściowym jest silniej zdelokalizowany niż w jonie wyjściowym, a większa polarność rozpuszczalnika spowalnia reakcję. Nawet w przypadku rozpuszczalników o mniej więcej tej samej polarności istnieje różnica między rozpuszczalnikami protonowymi i aprotonowymi. Reakcje SN1 niezjonizowanych substratów są szybsze w rozpuszczalnikach protonowych, które mogą tworzyć wiązania wodorowe z grupą opuszczającą. Wpływ polarności rozpuszczalnika na szybkość reakcji SN2 zależy od rozkładu ładunku w stanie przejściowym.
Podobny wpływ jak polarność rozpuszczalnika wywiera moc jonowa medium, w którym prowadzona jest reakcja. Ogólnie dodanie soli do roztworu w jakim prowadzona jest reakcja substytucji nukleofilowej wywiera taki skutek jak zwiększenie polarności rozpuszczalnika. Jednak gdy wprowadzana jest do roztworu sól zawierająca takie same jony jak grupa opuszczająca szybkość reakcji przebiegającej według mechanizmu SN1 ulega zmniejszeniu.
Nukleofile ambidentne: regioselektywność
Niektóre nukleofile mają parę elektronów na każdym z dwóch lub więcej atomów lub można rozrysować ich rezonansowe, w których dwa lub więcej atomów posiada pary elektronowe. W takich przypadkach nukleofil może zaatakować na dwa lub więcej różnych sposobów, dając różne produkty. Takie związki nazywane są nukleofilami ambidentami. W większości przypadków nukleofil z dwoma potencjalnie atakującymi atomami może, w zależności od warunków atakować jednym z nich, i często otrzymuje się mieszaniny produktów. Na przykład nukleofilowy NCO– zwykle daje tylko izocyjaniany RNCO, a nie izomeryczne cyjaniany ROCN. Gdy reakcja może potencjalnie spowodować powstanie dwóch lub więcej izomerów strukturalnych (np. ROCN lub RNCO), ale w rzeczywistości wytwarza tylko jeden mówi się, że reakcja jest regioselektywna. Niektóre ambidentne nukleofile to:
- Jony typu –CO–+CR–CO–; powstają w wyniku deprotonacji estrów malonowych, β-jetoestrów, β -diketonów; wykazują one struktury rezonansowe:
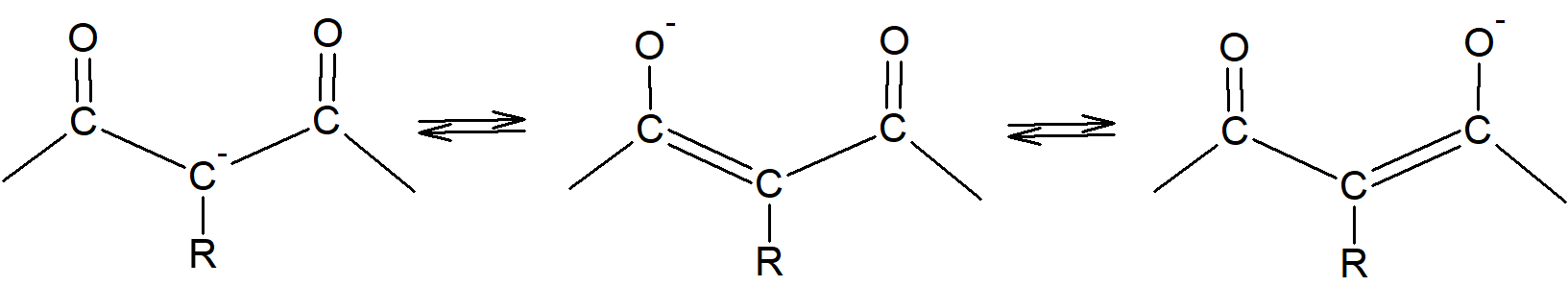
i mogą podlegać C- lub O-alkilowaniu:
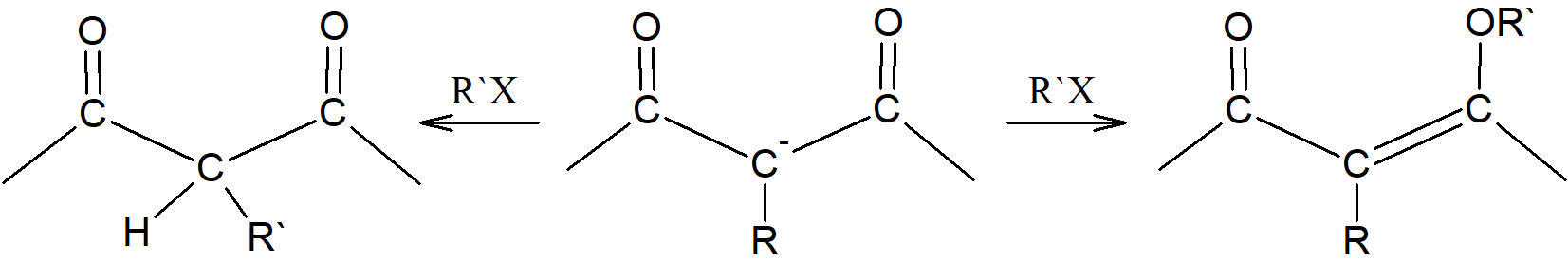
- Związki typu H3C–CO–CH2–CO– mogą odszczepiać w obecności silnej zasady dwa protony przekształcając się w H2C+–CO–+CH–CO–. Takie jony są ambidentami nukleofilami, a atak praktycznie zawsze zachodzi przez bardziej zasadowy węgiel. Ponieważ wodór w węglu związanym z dwiema grupami karbonylowymi jest bardziej kwaśny niż na węglu związanym tylko z jedną grupą CO, grupa CH jest zatem mniej zasadowa niż grupa CH2, więc ta ostatnia atakuje substrat.
- Inne przykłady jonów ambidentnych to jon CN–, który tworzy albo nitryle RCN lub izocyjanki RN≡C; jon nitrowy tworzącu estry azotynowe R–O–N=O lub związki nitrowe RNO2.
Podsumowując reakcję substytucji nukleofilowej można stwierdzić, że:
- Zgodnie z teorią twardych i miękkich kwasów i zasad twarde kwasy preferują twarde zasady, a miękkie kwasy miękkie zasady. W mechanizmie SN1 nukleofil atakuje karbokation, który jest twardym kwasem. W mechanizmie SN2 nukleofil atakuje atom węgla w cząsteczce, który jest bardziej miękkim kwasem. Bardziej elektroujemny atom ambidentnego nukleofila jest twardszą zasadą niż atom mniej elektroujemny. Możemy zatem stwierdzić, że gdy mechanizm danej reakcji zmienia się z SN1 na SN2, ambidentny nukleofil staje się bardziej podatny na atak ptzy mniej elektroujemnym atomie. Dlatego zmiana warunków reakcji prowadząca do zmiany mechanizmu z SN1 na SN2 powinna sprzyjać atakowi przez węgiel w przypadku CN–, atak przez azot w NO2–, oraz substytucję przy atomie węgla w jonach enolanowych. Przykładowo, pierwszorzędowe halogenki alkilowe są atakowane (w rozpuszczalnikach protonowych) przez atom węgla anionu CH3COCH2COOEt, podczas gdy α-chloroetery, które reagują zgodnie z mechanizmem SN1 mają nukleofilowy atom tlenu. Nie oznacza to jednak, że atak następuje przez atom mniej elektroujemny we wszystkich reakcjach SN2 i atom bardziej elektroujemny we wszystkich reakcjach SN1. Miejsce ataku zależy również od natury nukleofila, rozpuszczalnika, grupy opuszczającej i innych warunków. Można jedynie stwierdzić, że zwiększenie charakteru SN2 stanu przejściowego zwiększa prawdopodobieństwo ataku atomu mniej elektroujemnego.
- Wszystkie ujemnie naładowane nukleofile muszą oczywiście mieć dodatni przeciwjon. Jeśli jonem tym jest Ag+, a nie Na+ lub K+, to stan przejściowy ma charakter zbliżony do SN1. Dlatego zastosowanie Ag+ promuje atak na atom bardziej elektroujemny. Na przykład halogenki alkilowe traktowane NaCN generalnie dają głównie RCN, ale użycie AgCN zwiększa wydajność izocyjanków RNC.
- W wielu przypadkach rozpuszczalnik wpływa na pozycję ataku. Im mniej solwatowany nukleofil, tym bardziej prawdopodobne jest, że zaatakuje swoim atomem bardziej elektroujemnym, ale im bardziej ten atom jest otoczony cząsteczkami rozpuszczalnika lub dodatnimi przeciwjonami, tym większe jest prawdopodobieństwo ataku atomu o mniejszej elektroujemności. W rozpuszczalnikach protonowych atom bardziej elektroujemny jest lepiej solwatowany dzięki wiązaniom wodorowym niż atom mniej elektroujemny. W polarnych rozpuszczalnikach aprotonowych żaden atom nukleofila nie jest silnie solwatowany, ale rozpuszczalniki te są bardzo skuteczne w solwatowaniu kationów. Zatem w polarnym rozpuszczalniku aprotonowym bardziej elektroujemny koniec nukleofila jest mniej otoczony zarówno przez rozpuszczalnik, jak i kation, tak że zmiana z protycznego na polarny aprotonowy rozpuszczalnik często zwiększa zasięg ataku atomu bardziej elektroujemnego. Zmiana kationu z Li+ na Na+ lub K+ ( w rozpuszczalnikach niepolarnych) faworyzuje O- alkilowanie (K+ znacznie słabiej oddziałuje z nukleofilem niż Li+).
- W skrajnych przypadkach efekty steryczne mogą sterować regioselektywnością reakcji.
Substytucja nukleofilowa w pierścieniu aromatycznym
Najważniejszy mechanizm nukleofilowej substytucji aromatycznej składa się z dwóch etapów, ataku cząsteczek nukleofilowych na węgiel ipso pierścienia aromatycznego (węgiel przy którym znajduje się grupa opuszczająca), po którym następuje eliminacja grupy opuszczającej i regeneracja pierścienia aromatycznego.
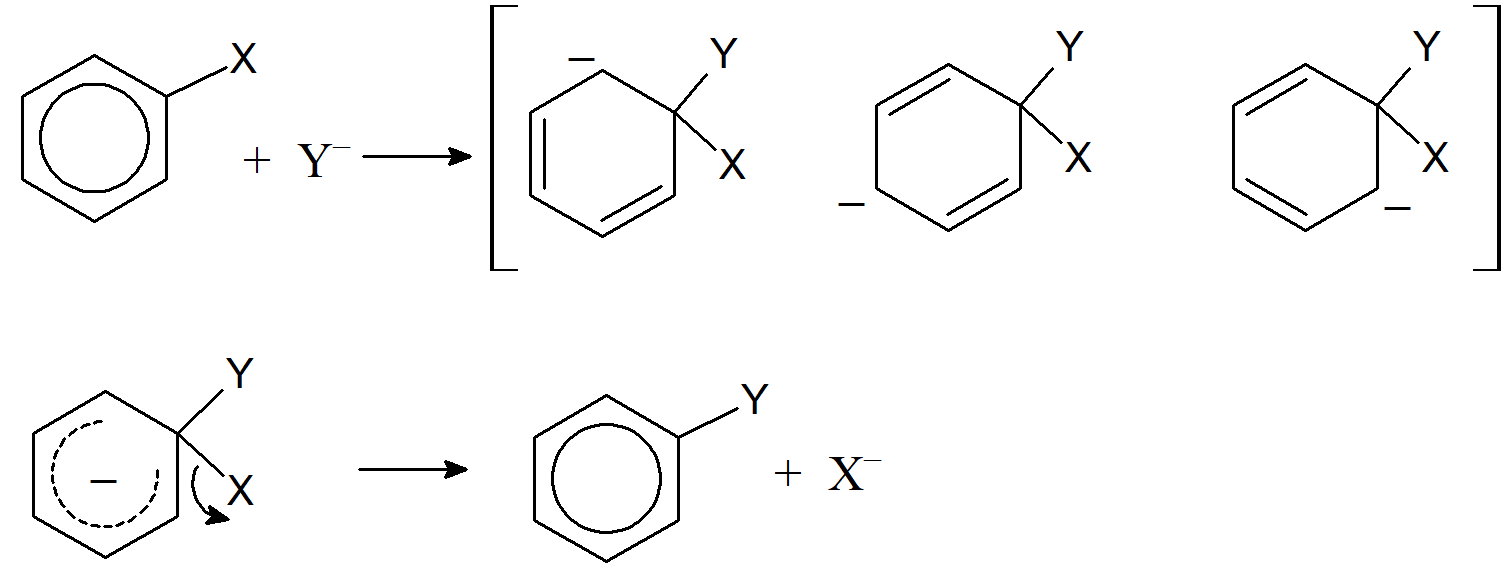
Pierwszy etap najczęściej determinuje szybkość reakcji.
Mechanizm jednocząsteczkowy SN1 występuje bardzo rzadko i odgrywa rolę praktycznie jedynie w przypadku soli diazoniowych, gdzie zachodzące procesy można przedstawić w postaci:
Ar-N≡N ↔ Ar+ + N2
Ar+ + Y– → Ar–Y
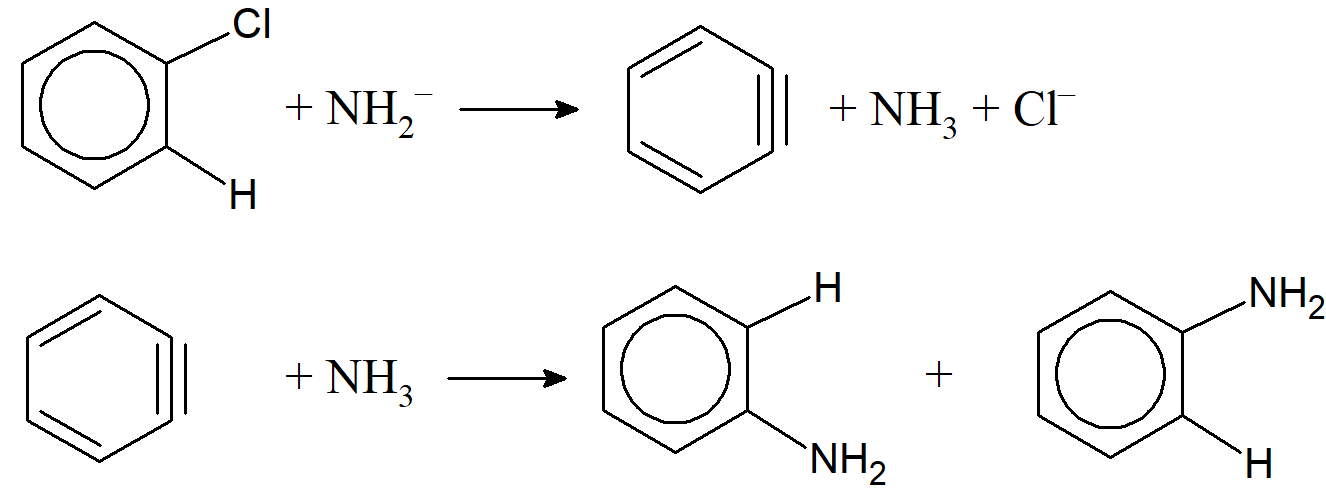
Kolejnym mechanizmem jest mechanizm arynowy, który odgrywa rolę w przypadku halogenopochodnych arenowych i obejmuje etap eliminacji po którym następuje addycja.