 Principles of Chemistry
Principles of Chemistry
 Mechanizmu reakcji związków nieorganicznych
Mechanizmu reakcji związków nieorganicznych
Reakcje z udziałem związków nieorganicznych, jak i znacznej liczby związków koordynacyjnych, są reakcjami szybkimi, a powstające produkty charakteryzują się trwałością termodynamiczną w odróżnieniu od związków organicznych gdzie często dominuje trwałość kinetyczna.
Wszystkie reakcje chemiczne mają ograniczoną szybkość co wynika z faktu, że cząsteczki reagujących substancji musza się do siebie zbliżyć aby reakcja mogła zajść. Dyfuzja jest bardzo szybka w gazach i cieczach i powolna w fazie stałej. Jednak tak w gazach jak i w cieczach szybkość reakcji jest mniejsza niż dyfuzja co jest związane z energią aktywacji reakcji. Można to przedstawić graficznie w sposób następujący:
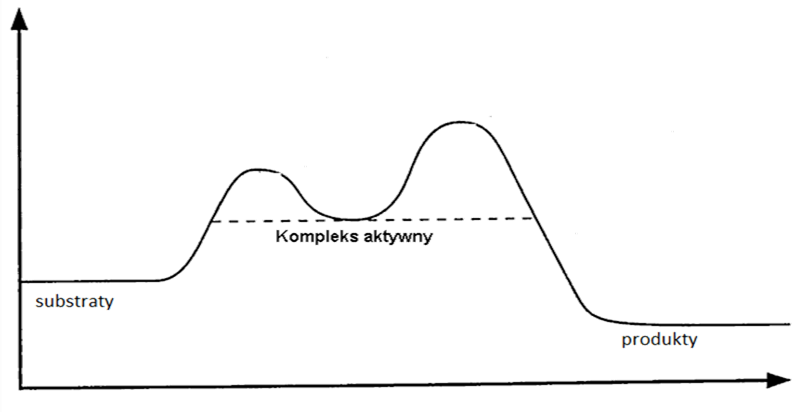
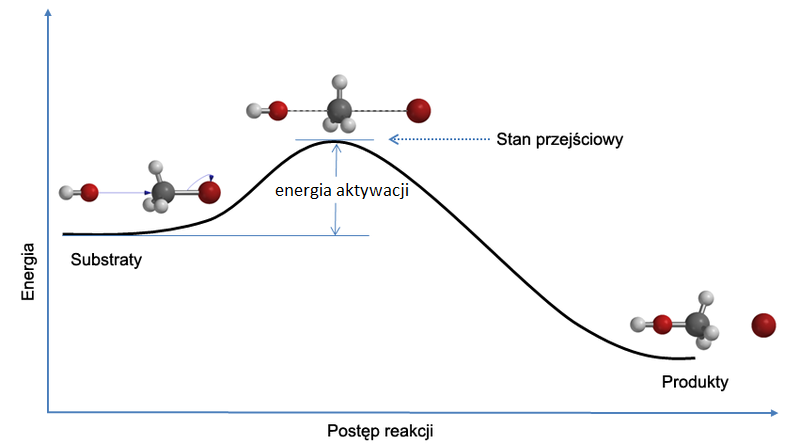 Zakłada się, że w reakcji występuje wzbudzony stan przejściowy, w którym położenia jąder substratów zostają zmienione w taki sposób, że sprzyja to bezpośrednio powstani9u produktów. Im niższa energia aktywacji tym łatwiej jest osiągany stan przejściowy i szybsza reakcja. W rzeczywistości istnienie stanu przejściowego jest mało prawdopodobne gdyż należałoby mu przypisać maksymalną energię potencjalną. Jednak rozważanie takiej struktury jest celowe gdyż można ocenić na ile prawdopodobna, od strony energetycznej, byłaby jego struktura. Z drugiej strony stan pośredni w reakcji dwustopniowej posiada minimum energii, która pomimo tego, że jest wysoka to jednak znajduje się w minimum studni potencjału i w tym przypadku można go wykryć doświadczalnie.
Zakłada się, że w reakcji występuje wzbudzony stan przejściowy, w którym położenia jąder substratów zostają zmienione w taki sposób, że sprzyja to bezpośrednio powstani9u produktów. Im niższa energia aktywacji tym łatwiej jest osiągany stan przejściowy i szybsza reakcja. W rzeczywistości istnienie stanu przejściowego jest mało prawdopodobne gdyż należałoby mu przypisać maksymalną energię potencjalną. Jednak rozważanie takiej struktury jest celowe gdyż można ocenić na ile prawdopodobna, od strony energetycznej, byłaby jego struktura. Z drugiej strony stan pośredni w reakcji dwustopniowej posiada minimum energii, która pomimo tego, że jest wysoka to jednak znajduje się w minimum studni potencjału i w tym przypadku można go wykryć doświadczalnie.
 Ponieważ powstanie stanu przejściowego łączy się ze zmianami długości wiązań w cząsteczkach substratów, a nawet z ich zrywaniem lub powstawaniem nowych, rozpatrując mechanizm reakcji można skupić się na dwóch zlokalizowanych wiązaniach, które zostają zerwane lub utworzone, albo rozważać zmiany położenia wszystkich atomów w cząsteczce opisując oscylacyjny stan przejściowy. Pierwsze podejście jest prostsze, a drugie bardziej ogólne, ale w obydwu przypadkach szacowana jest bariera energetyczna tworzeni9a stanu przejściowego.
Ponieważ powstanie stanu przejściowego łączy się ze zmianami długości wiązań w cząsteczkach substratów, a nawet z ich zrywaniem lub powstawaniem nowych, rozpatrując mechanizm reakcji można skupić się na dwóch zlokalizowanych wiązaniach, które zostają zerwane lub utworzone, albo rozważać zmiany położenia wszystkich atomów w cząsteczce opisując oscylacyjny stan przejściowy. Pierwsze podejście jest prostsze, a drugie bardziej ogólne, ale w obydwu przypadkach szacowana jest bariera energetyczna tworzeni9a stanu przejściowego.
Entropia aktywacji
Energia aktywacji na ogół określa przebieg reakcji chemicznej, jednak nie zawsze tak jest. Z teorii stanu przejściowego wyrażenie na stałą szybkości reakcji ma postać:
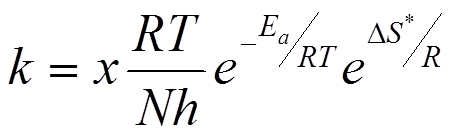 gdzie x oznacza współczynnik przejścia, przyjmowany z reguły za 1 gdyż jego wartość jest trudna do wyznaczenia, R /N stałą Boltzmanna, Ea energię aktywacji Arrheniusa, ΔS* entropię aktywacji, h stałą Plancka.
gdzie x oznacza współczynnik przejścia, przyjmowany z reguły za 1 gdyż jego wartość jest trudna do wyznaczenia, R /N stałą Boltzmanna, Ea energię aktywacji Arrheniusa, ΔS* entropię aktywacji, h stałą Plancka.
Entalpie aktywacji reakcji w roztworze określa zależność: ΔH* = Ea – RT, a entropia aktywacji jest równa różnicy entropii substratów i entropii stanu przejściowego. W związku z tym można oczekiwać, że entropia tworzenia stanu przejściowego, w którym następuje łączenie dwóch cząsteczek substratu, będzie mniejsza niż entropia stanu przejściowego, w którym następuje dysocjacja. Problemem jest fakt, że w roztworach występują efekty solwatacji, które w decydujący sposób mogą wpływać na zmiany entropii zwłaszcza gdy stan przejściowy jest bardzie spolaryzowany niż substraty. Chociaż rozważania reakcji związków nieorganicznych często opierają się na energii aktywacji to zmiany entropii mogą mieć decydujące znaczenie. Przykładowo reakcja pomiędzy [Au(dien)Cl]2+ i metanolem zachodzi 30 000 razy szybciej niż analogiczna reakcja izoelektronowego i izostrukturalnego [Pt(dien)Cl]+. W obydwu reakcjach wartości entalpii aktywacji reakcji są zbliżone, a różnice w szybkości są związane z różnicami w entropii aktywacji tych reakcji.
Tworzenie i zrywanie wiązań
Opierając się na modelu wiązań zlokalizowanych w teorii orbitali cząsteczkowych można te procesy opisać w oparciu o dwa schematy:
- każdy atom biorący udział w wiązaniu zatrzymuje jeden elektron w wyniku czego powstają dwa rodniki, z których każdy zawiera jeden niesparowany elektron, czyli powstają dwa rodniki i mamy do czynienia z rozpadem homolitycznym;
- para elektronowa tworząca wiązanie pozostaje przy jednym z atomów. Taki proces nosi nazwę rozpadu heterolitycznego.
Prawda jest, że rozpad heterolityczny jest bardziej prawdopodobny dla związków polarnych (jonowych), ale w przypadku związków takich jak H3NBF3, gdzie wiązanie pomiędzy azotem i borem powstaje w wyniku przeniesienia pary elektronowej azotu na akceptorowy orbital boru, rozpad wiązania prowadzi do utworzenia obojętnych cząsteczek amoniaku i fluorku boru.
Tworzenie wiązań jest procesem stosunkowo prostym. Otóż układy rodnikowe, posiadające niesparowane elektrony łączą się ze sobą tworząc typowe, dwuelektronowe, wiązanie. Jednak w przypadku związków nieorganicznych zachowanie rodników nie jest tak proste. Przykładowo jon Mn(II) jest układem posiadającym pięć niesparowanych elektronów, ale trudno traktować go jako rodnik w potocznym rozumieniu i nie przejawia on skłonności do uczestnictwa w reakcjach o mechanizmie rodnikowym. Generalnie heterolityczne tworzenie wiązania, w związkach nieorganicznych, polega na przeniesieniu pary elektronowej od jednego atomu do drugiego, czyli jest to reakcja pomiędzy kwasem i zasadą Lewisa.
W reakcjach tworzenia wiązań substancje dzieli na dwie grupy:
- nukleofile czyli takie, które wykazują zdolność do reagowania z centrami dodatnimi i zachowują się jak donory elektronów;
- elektrofile czyli te, które wykazują skłonność do przyjmowania elektronów i reakcji z ujemnymi centrami.
W związku z takim podziałem przyjmujemy, że zasady Lewisa są nukleofilami, a kwasy Lewisa elektrofilami. Ponieważ elektrofilowość i nukleofilowośc jest oceniana na podstawie szybkości reakcji to pojęcia te nie mają nic wspólnego ze skalą zasadowości. Jest to jeden z powodów powstania koncepcji miękkich i twardych kwasów i zasad. Przykładowo jon jodkowy jest słabą zasadą ale bardzo dobrą substancją nukleofilową. Dodatkowo nukleofilowość i elektrofilowość silnie zależą od rozpuszczalnika. W szeregu przypadków kierunek reakcji i rodzaj produktów można stosunkowo łatwo przewidzieć opierając się na rozkładzie ładunków w cząsteczkach substratów, zakładając, że najbardziej nukleofilowy atom jednej cząsteczki połączy się z najbardziej elektrofilowym atomem drugiej. O ile takie podejście dobrze sprawdza się w chemii organicznej, to w chemii związków nieorganicznych czasami trudno ocenić, który atom (fragment) cząsteczki jest nukleofilowy czy elektrofilowy.
Kolejną trudność jaką napotyka koncepcja tworzenia i zrywania wiązań w reakcjach chemicznych związków nieorganicznych, jest wyjaśnienie mechanizmu tworzenia czy zrywania wiązań kowalencyjnych. Dodatkowo sytuację komplikuje fakt, że w reakcjach związków nieorganicznych tworzeniu lub zrywaniu ulega kilka wiązań. Przykładowo wprowadzenie wodoru do cząsteczki związku może być związane z równoległym utworzeniem dwóch wiązań σ.
Na gruncie teorii orbitali cząsteczkowych mamy dwie możliwości zerwania wiązań chemicznych – albo elektrony będą usuwane z orbitali wiążących albo są wprowadzane na cząsteczkowy orbital antywiążący. Pomijając wzbudzenie, czyli przeskok elektronu pomiędzy orbitalami, proces taki może zostać zrealizowany albo w wyniku zmieszania zapełnionego orbitalu cząsteczkowego z pustym orbitalem, lub też zmieszania pustego orbitalu antywiążącego z obsadzonym orbitalem cząsteczkowym. Prostym przykładem takiej reakcji jest wymiana chloru pomiędzy cząsteczką kwasu solnego a jonem chlorkowym:
Cl– + HCl → ClH + Cl–
Schematycznie można przedstawić ten proces następująco:
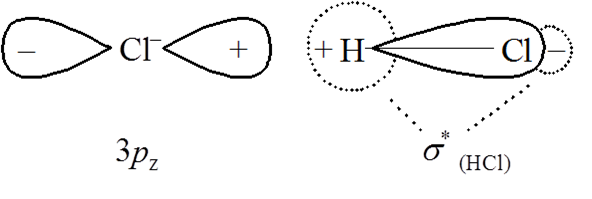 W reakcji kwasu i zasady Lewisa zbliżanie się cząsteczki zasady do cząsteczki kwasu powoduje wzrost nakładania orbitali donora i akceptora w wyniku czego tworzy się obsadzony orbital cząsteczkowy. Nukleofilowy jon chlorkowy zbliża się do cząsteczki HCl, a jego donorowy orbital pokrywa się z antywiążącym orbitalem cząsteczkowym kwasu, który jest zlokalizowany głównie na atomie wodoru. Tworzy się nowe wiązanie spowodowane obsadzeniem antywiążącego orbitalu σ w cząsteczce HCl, a następnie zachodzi odszczepienie jonu chlorkowego powstającego z pierwotnie kowalencyjnego wiązania w cząsteczce HCl. Inaczej mówiąc powstanie wiązania Cl–H powoduje osłabienie wiązania H–Cl. Pośredni związek postaci Cl–H–Cl został wyizolowany i stwierdzono, że odległość Cl–Cl jest znacznie większa niż dwukrotna odległość H–Cl.
W reakcji kwasu i zasady Lewisa zbliżanie się cząsteczki zasady do cząsteczki kwasu powoduje wzrost nakładania orbitali donora i akceptora w wyniku czego tworzy się obsadzony orbital cząsteczkowy. Nukleofilowy jon chlorkowy zbliża się do cząsteczki HCl, a jego donorowy orbital pokrywa się z antywiążącym orbitalem cząsteczkowym kwasu, który jest zlokalizowany głównie na atomie wodoru. Tworzy się nowe wiązanie spowodowane obsadzeniem antywiążącego orbitalu σ w cząsteczce HCl, a następnie zachodzi odszczepienie jonu chlorkowego powstającego z pierwotnie kowalencyjnego wiązania w cząsteczce HCl. Inaczej mówiąc powstanie wiązania Cl–H powoduje osłabienie wiązania H–Cl. Pośredni związek postaci Cl–H–Cl został wyizolowany i stwierdzono, że odległość Cl–Cl jest znacznie większa niż dwukrotna odległość H–Cl.
Z teorii orbitali cząsteczkowych wynika, że dwa dostępne elektrony , w kompleksie przejściowym muszą wytworzyć dwa wiązania Dodatkowo oscylacje cząsteczki kompleksu aktywnego umożliwiają obniżenie symetrii co powoduje zmieszanie orbitali produktów i wytworzenie orbitali produktów. W ten sposób można stwierdzić, że prawdopodobieństwo zajścia reakcji jest uzależnione od nakładania się odpowiednich orbitali. A jeżeli nie jest to możliwe to od drgań cząsteczki, które umożliwią nakładanie się orbitali substratów. To prowadzi do konieczności uwzględnienia symetrii orbitali w reakcjach chemicznych.
Symetria w reakcjach chemicznych
Rozważanie symetrii pod katem jej zmian w reakcjach chemicznych nie należy do prostych zagadnień. Przede wszystkim należy zdać sobie sprawę z faktu, że w czasie przebiegu reakcji chemicznej nie jest możliwa zmiana symetrii względem operacji symetrii jakim podlegają orbitale cząsteczkowe. Innymi słowy symetria ulega "zamrożeniu" ponieważ jakakolwiek oscylacja związana z reakcją czy też mieszaniem orbitali musi być pełnosymetryczna czyli typu A1 względem elementów symetrii zachowanych w trakcie reakcji. Przykładowo w przytoczonej powyżej reakcji drganie ruchów jąder polega na asymetrycznym rozciąganie co powoduje, że symetria cylindryczna układu zostaje zachowana, a samo drganie jest symetryczne ze względu na tą symetrię. Innym przykładem może być reakcja pomiędzy fluorem i bromem.
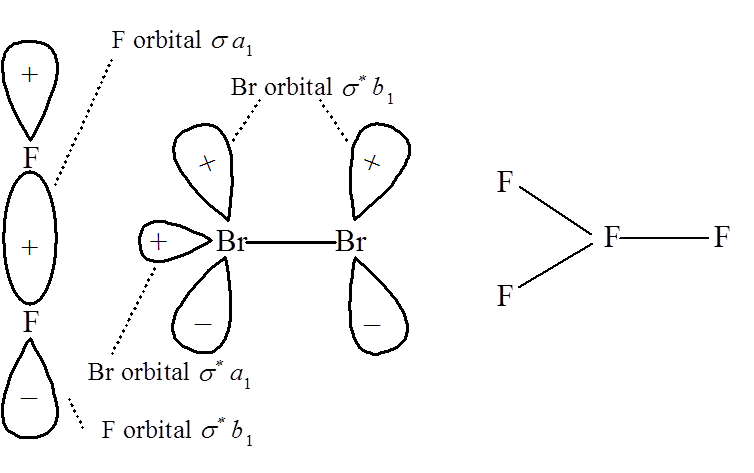 Antywiążące orbitale typu π* cząsteczki Br2 mogą nakładać się na orbitale σ* F2 ponieważ obydwa są typu b1. Dodatkowo orbitale σ* obydwu cząsteczek typu a1 mogą tworzyć wiążące orbitale cząsteczkowe Br–F. Reakcja jest dozwolona symetrią, chociaż produkt Br2F2 jest wysoce reaktywny.
Antywiążące orbitale typu π* cząsteczki Br2 mogą nakładać się na orbitale σ* F2 ponieważ obydwa są typu b1. Dodatkowo orbitale σ* obydwu cząsteczek typu a1 mogą tworzyć wiążące orbitale cząsteczkowe Br–F. Reakcja jest dozwolona symetrią, chociaż produkt Br2F2 jest wysoce reaktywny.
Bardziej skomplikowane przykłady dotyczą chemii organicznej i są związane z otwarciem pierścienia cyklobutenu. Rozerwanie wiązań C–C wymaga zerwania wiązania typu σ i wiązania π. W takim przypadku, dla symetrii C2V symetrie obsadzonych orbitali substratu i produktów nie odpowiadają sobie i dlatego nie można oczekiwać osłabienia wiązania w wyniku zmieszania obsadzonego z pustym orbitalem cząsteczkowym. Inaczej mówiąc symetria C2V nie może zostać zachowana podczas tej reakcji. Jednak wniosek ten jest prawidłowy jedynie gdy uznamy, że atomu wodoru grup metylenowy w cyklobutenie musza być przesunięte poza płaszczyznę atomów węgla. Taka operacja jest niemożliwa przy zachowaniu symetrii C2V w butadienie. Można zachować albo oś dwukrotną albo płaszczyznę symetrii xz. Rozważenie symetrii cyklobutanu i butadienu wskazuje, że otwarcie pierścienia zachodzi z zachowaniem osi dwukrotnej, co potwierdzają badania z użyciem izotopowo znaczonych atomów wodoru.
Rozpatrzenie symetrii pozwala na określenie prawdopodobieństwa mieszania się orbitali, ale stopień ich wzajemnego oddziaływania jest uzależniony od różnicy energii pomiędzy nimi. Założenie tow, wynikające z rachunku zaburzeń, stanowi podstawę teorii orbitali granicznych (Teoria Fukui), zgodnie z którą reakcja chemiczna przebiega w kierunku i miejscu maksymalnego nakładania się obsadzonych orbitali cząsteczkowych o najwyższych energiach i nieobsadzonych orbitali cząsteczkowych o najniższych energiach w cząsteczkach substratów. Zasada ta jest dobrze spełniona w przypadku reakcji zachodzących w cząsteczkach organicznych. Natomiast w chemii nieorganicznej napotyka ona trudności, z których główną jest problem zdefiniowania orbitali granicznych cząsteczki. Cząsteczki związków nieorganicznych charakteryzują się często, zbiorami orbitali o zbliżonych energiach, a dodatkowo często orbitale zbliżone pod względem energii do orbitali granicznych odgrywają decydującą rolę w reakcjach cząsteczek nieorganicznych. Istotne jest również to, aby symetrie orbitali substratów i produktów korelowały ze sobą.
Przykładem trudności jakie napotykamy przy analizę mechanizmów reakcji związków nieorganicznych może być prosta reakcja tworzenia jodowodoru. Przez długi czas uważano, że jest to dwucząsteczkowa reakcja ze stanem przejściowym przedstawionym na poniższym schemacie.
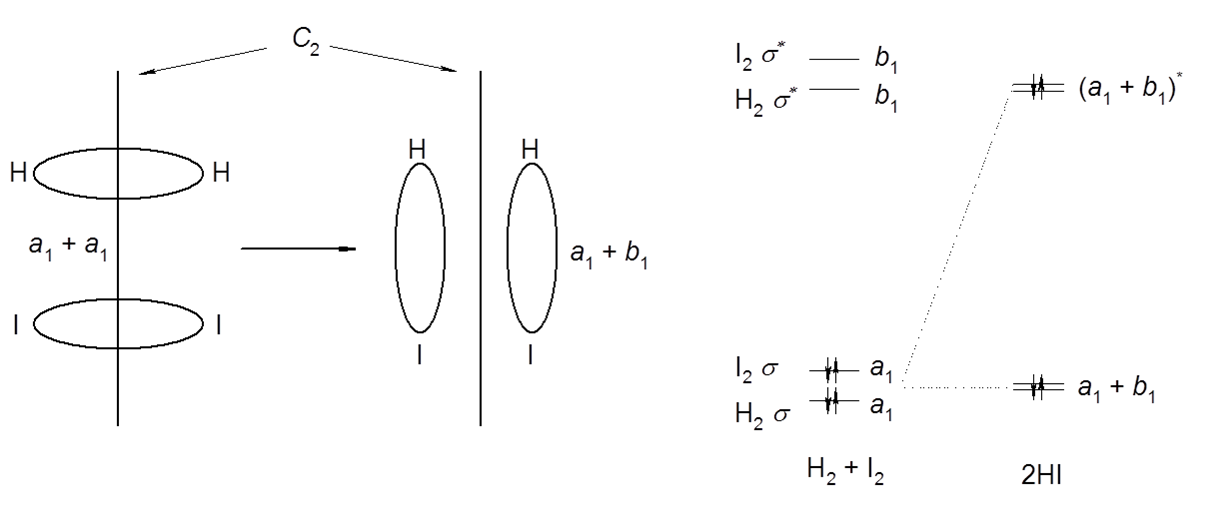 Ze schematu wynika, że pomimo możliwości oddziaływania orbitali a1 to produkty tworzą się z udziałem obsadzonego antywiążącego orbitalu a1*. Taki stan wymaga dostarczenia energii, a powstający produkt jest nietrwały. Ponieważ widać, że symetrie zrywanych i tworzonych wiązań w tej reakcji są takie same, to rzeczywisty przebieg tej reakcji nie wymaga powstawania wzbudzonego stanu pośredniego.
Ze schematu wynika, że pomimo możliwości oddziaływania orbitali a1 to produkty tworzą się z udziałem obsadzonego antywiążącego orbitalu a1*. Taki stan wymaga dostarczenia energii, a powstający produkt jest nietrwały. Ponieważ widać, że symetrie zrywanych i tworzonych wiązań w tej reakcji są takie same, to rzeczywisty przebieg tej reakcji nie wymaga powstawania wzbudzonego stanu pośredniego.
Podsumowując można stwierdzić, że:
- tworzenie i zrywanie wiązań wynika z wzajemnego oddziaływania orbitali atomowych lub cząsteczkowych w centrach reakcji. Mieszanie się orbitali może być wynikiem albo zbliżania się do siebie cząsteczek substratów, lub też być wynikiem odkształcenia geometrii cząsteczki wynikającego z oscylacji (efekt Jahna-Tellera drugiego rzędu).
- Wzajemne oddziaływanie orbitali będzie największy gdy ich energie będą zbliżone.
- Obsadzone orbitale substratów i produktów powinny korelować ze sobą co jest warunkiem przegrupowania elektronów w czasie reakcji.
Mechanizmy reakcji, w których utrudnione jest mieszanie się orbitali lub brak jest korelacji między nimi są związane z większymi energiami aktywacji. Kryterium mieszania orbitali jest najbardziej istotne w przypadku gdy powstające wiązania mają charakter kowalencyjny. Obojętne, niepolarne cząsteczki nie będą ze sobą silnie oddziaływały bez zmieszania odpowiednich orbitali. Inaczej sytuacja wygląda w przypadku układów jonowych lub silnie polarnych. W tych przypadkach potencjały elektrostatyczne cząsteczek mogą na tyle silnie zmieniać energię układu, że powstaniu wiązań nie musi towarzyszyć mieszanie orbitali. Inaczej mówiąc rozpatrywanie reakcji układów jonowych na gruncie mieszania się orbitali reagujących cząsteczek jest pozbawione sensu.
Istotnym warunkiem określającym reakcje jest wymaganie aby w trakcie reakcji całkowity spin nie ulegał zmianie. Zmiana spinu w trakcie reakcji powoduje zmniejszenie szybkości reakcji, ale uwzględnienie sprzężenia spinowo-orbitalnego powoduje, że reakcje takie zachodzą z normalną szybkością.