 Principles of Chemistry
Principles of Chemistry
 Struktura atomu
Struktura atomu
Współczesna teoria struktury atomowej (szerokie opracowanie tego zagadnienia jest dostępne pod tym linkiem) opiera się na odkryciach elektronu i radioaktywności. Promienie katodowe wytwarzane przez wyładowania elektryczne w gazach pod niskim ciśnieniem zostały odkryte przez J. Plūckera w 1859 r., i zbadane przez J. W. Hittorfa (1869), W. Crookesa (1876 itd.) i J. J. Thomsona. Ten ostatni wykazał w 1897 r., że są to ujemnie naładowane cząstki o masie znacznie mniejszej niż masa atomu wodoru. Przypuszczał, że cząstki te są składnikami wszystkich atomów i nazwał je „ciałkami”. Nazwa elektron została zaproponowana przez Johnstona Stoneya (który obliczył przybliżoną wartość jego ładunku w 1874 r.).
Radioaktywność uranu odkrył H. Becquerel w 1896 roku. W 1898 roku Pierre Curie, M. Skłodowska-Curie. i G. Bémont uzyskali z blendy uranowej radioaktywne pierwiastki: polon i rad, a radioaktywność toru została odkryta niezależnie przez G. C. Schmidta i M. Skłodowską-Curie w 1898 r. W 1899 r. M. Skłodowska-Curie zasugerowała, że radioaktywne atomy są niestabilne i rozpadają się wraz z emisją energii, hipoteza ta została rozszerzona przez E. Rutherforda i F. Soddy'ego w 1903 roku. F. O. Giesel, H. Becquerel, Piotr i Maria Skłodowska-Curie oraz E. Rutherford w latach 1899–1900 zidentyfikowali promieniowanie α, β i γ emitowane przez substancje radioaktywne. W 1901 r. F. E. Dorn odkrył emanację radu, a w 1904 r. W. Ramsay i F. Soddy wykazali, że emanacja ta zmienia się samorzutnie w hel. E. Regener i E. Rutherford udowodnili, że promienie α są dodatnio naładowanymi atomami helu.
Atomy radu i toru podlegają kolejno zmianom, z których każdy charakteryzuje się określoną szybkością rozpadu, przy czym niektóre etapy pośrednie mają bardzo krótki czas życia. Produktem końcowym rozpadu zarówno radu, jak i toru jest ołów. Natomiast rad jest jednym z produktów rozpadu uranu. Na podstawie liczby emitowanych cząstek α, o masie 4, okazało się, że ciężary atomowe dwóch rodzajów ołowiu powinny być różne: ołów pochodzący z uranu ma masę 206, a ołów pochodzący z rozpadu toru powinien mieć masę 208, natomiast w przyrodzie występuje ołów o masie atomowej 207. Potwierdził te F. Soddy, i niezależnie T. W. Richards, w 1913 roku, Soddy nazwał różne odmiany tego samego pierwiastka izotopami.
W 1911 r. F. Soddy zwrócił uwagę, że emisja promieniowania a powoduje powstania pierwiastka znajdującego się o dwa miejsca wcześniej w układzie okresowym niż wyjściowy, a w 1913 r. A. S. Russell i K. Fajans uogólnili to spostrzeżenie uwzględniając promieniowanie β, którego emisja daje pierwiastek leżący o jedno miejsce dalej niż wyjściowy. Tak zwane prawo przesunięć reguluje pozycje w układzie okresowym pierwiastków promieniotwórczych i produktów ich rozpadu. Ponieważ wszystkie one mają tę samą liczbę atomową, to wszystkie izotopy danego pierwiastka zajmują to samo miejsce w układzie okresowym.
Istnienie izotopów pierwiastków nie promieniotwórczych odkrył J. J. Thomson w 1913 r. W przypadku neonu, który został odkryty, metodą polegającą na rozdzielaniu za pomocą pól magnetycznych i elektrycznych tak zwanych promieni dodatnich wytwarzanych przez wyładowania elektryczne w gazach, okazało się, że składa się on z mieszaniny dwóch rodzajów atomów o masach 20 i 22, teraz oznaczonych jako 20Ne i 22Ne. Modyfikacja tej samej metody zastosowana przez F. Astona wykazała, że większość pierwiastków to mieszaniny izotopów, np. chlor to 35C1 i 37C1, przy czym masa atomowa 35,46 jest wartością średnią i atom o takiej niecałkowitej masie nie występuje. Izotopy tlenu, azotu, węgla itp. zostały wykryte metodami spektroskopowymi, przy czym najbardziej uderzającym odkryciem dokonanym przez H. C. Ureya z Columbia University w 1932 r. był izotop wodoru o masie 2, czyli deuter.
W 1911 r. E. Rutherford stwierdził, że bardzo duże ugięcia, jakich czasami doświadczają cząstki α podczas przechodzenia przez materię, można wyjaśnić, jeśli zakłada się, że każdy atom składa się z bardzo małego dodatnio naładowanego jądra otoczonego elektronami zewnętrznymi w stosunkowo dużych odległościach od jądro. Cząstka α składałaby się wówczas z jądra helu i chociaż normalnie może przechodzić przez zewnętrzne części innego atomu, jest odchylana, gdy zbliża się do małego jądra o ładunku dodatnim. Jądro atomu wodoru nazwano protonem, mającym ładunek równy, ale przeciwny do ładunku elektronu i masę w przybliżeniu taką samą jak atom wodoru. Jądra innych atomów powinny zatem składać się z protonów i neutronów, przy czym neutron jest cząstką o, w przybliżeniu, tej samej masie co proton, ale pozbawiona ładunku. Masa atomu jest w przybliżeniu równa sumie mas protonów i neutronów; ładunek jądrowy jest równy liczbie protonów (i tym samym elektronów). Związek między liczbą atomową a ładunkiem jądrowym został wyjaśniony w 1913-14 przez H. Moseleya, pracującego w Manchesterze i Oksfordzie, który stwierdził, że każdy pierwiastek bombardowany promieniami katodowymi emituje promieniowanie rentgenowskie o charakterystycznej częstotliwości, ν , zależnej od liczby atomowej, N, i założył, że N jest równe ładunkowi dodatniemu zlokalizowanemu w jądrze. Długość fali różniła się to o jedną jednostkę, przechodząc od jednego pierwiastka do następnego w układzie okresowym. W szeregu liczb atomowych znaleziono pewne luki, ale zostały one wypełnione odkryciem brakujących pierwiastków, takich jak hafn odkryty przez D. Costera i G. von Hevesy'ego w 1923 r. oraz ren odkryty przez W. Noddacka. I. Tocke i O. Berga w 1025 r. Metoda Moseleya pozwoliła na zbadanie pierwiastków i wykazała, że, z kilkoma wyjątkami, układ okresowy był kompletny.
Teorię atomową znacznie rozszerzył w 1913 r. Niels Bohr, profesor w Kopenhadze, który wykazał, że model atomowy Rutherforda, dzięki wprowadzeniu teorii kwantowej Plancka, może wyjaśnić widma pierwiastków, a także ich pozycję w układzie okresowym. Prosty model atomu zaproponował w 1916 r. G. N. Lewis, profesor z Berkeley w Kalifornii, który przypuszczał, że zewnętrzna warstwa elektronowa atomów gazów szlachetnych wynosi 8, a atomy pozostałych pierwiastków dążą do uzyskania takiej właśnie konfiguracji powłoki zewnętrznej poprzez przyjęcie lub utratę zewnętrznych elektronów, tworząc jony ujemne lub dodatnie (jak sugerowane przez A. Kossel w 1916 r.) lub poprzez współdzielenie par elektronów z innymi atomami, tworząc wiązania walencyjne. Teorię rozszerzył na atomy cięższych pierwiastków Irving Langmuir w 1919 r.
W teorii atomu Bohra elektrony były uważane za ładunki punktowe krążące wokół centralnego dodatniego jądra atomu na orbitach kołowych. Stan energetyczny elektronu oznaczono liczbą kwantową n, przy czym każda orbita ma określoną wartość liczby n. Analogicznie do orbit planetarnych można by oczekiwać, że orbity będą elipsami, a dla danej wartości n elipsy o różnej mimośrodowości są możliwe. A. Sommerfeld (1915 r.) wprowadził kolejną liczbę kwantową l w celu uwzględnienia tej cechy, a późniejsza teoria wykazała, że l jak n mają wartości całkowite, z których najwyższa wartość l jest o jeden mniejsza niż n, np. jeśli n = 4, możliwe wartości l wynoszą 3, 2, 1, 0. Jeśli płaszczyzna elipsy jest związana z pewnym słabym polem magnetycznym, może ona przyjąć pewną liczbę dyskretnych pozycji, które są zdefiniowane przez inną liczbę kwantową m, która ma wartości równe dodatnim i ujemnym wartościom l. Czwarta liczba kwantowa s określa spin elektronu mający dwie wartości ±½ odpowiadające dwóm przeciwnym kierunkom spinu; tak zwana „zasada wykluczenia” określona przez Pauliego (1925 r.) twierdzi, że w dowolnym atomie nigdy nie może istnieć więcej niż jeden elektron o danym zestawie wszystkich czterech liczb kwantowych n, m, l i s. Przyjmując wartości 1, 2, 3 i 4 dla n i wykorzystując powyższe zależności, okazuje się, że maksymalna liczba elektronów o tych wartościach n wynosi 2, 8, 18 i 32. Są to liczby pierwiastków w okresach układu okresowego:
n = I, stąd l = 0 i m = 0 ; s = ±½ co odpowiada 2 elektronom.
n = 2, stąd l wynosi 1 lub 0. Dla l = 0, m = 0; dla l = 1, m wynosi –1, 0 lub 1.
Istnieją cztery wartości m i dla każdego dwie wartości s dają w sumie 8 możliwych elektronów.
n = 3, stąd l wynosi 2, 1 lub 0; dla tych trzech przypadków m wynosi (2, 1, 0, –1, –2), (1, 0, –1) i 0; istnieje 9 możliwych przypadków, dla każdego s = ±½, co daje w sumie 18.
n = 4, stąd l wynosi 3, 2, 1 lub 0, co daje 16 wartości m, i 32 w sumie.
Struktury elektronowe atomów wszystkich pierwiastków znane są na podstawie danych spektroskopowych. Chociaż model atomowy Bohra z elektronami krążącymi na orbitach nie jest już akceptowany, znaczenie liczb kwantowych pozostaje aktualne. Wystarczy uznać elektrony za rozmieszczone w „powłokach” odpowiadających specjalnym wartościom n, przy czym maksymalna liczba elektronów w tych powłokach, zaczynając od tej najbliżej jądra, wynosi 2, 8, 18 i 32. Koniec okresu w układzie zamyka gaz szlachetny, a liczba elektronów na zewnętrznej powłoce tego gazu wynosi 2 dla helu i 8 dla wszystkich pozostałych.
Jądro atomowe zbudowane jest z protonów i neutronów, przy czym liczba protonów określa dodatni ładunek jądra lub liczbę atomową. Liczba elektronów, znajdujących się poza jądrem, jest taka aby atom był obojętny, czyli odpowiada dodatniemu ładunkowi jądra. Elektrony mają główne liczby kwantowe n wynoszące 1, 2, 3 itd. Dla n = 1 zasada Pauliego pokazuje, że możliwe są tylko dwa takie elektrony; atom wodoru składa się z protonu i jednego elektronu, a atom helu z jądra o dwóch protonach i dwóch neutronach (liczba atomowa 2) i dwóch elektronów. Ten pierwiastek kończy pierwszy okres złożony z dwóch pierwiastków. Następne dodawane elektrony mają n = 2, a zasada Pauliego pokazuje, że może być ich osiem, odpowiadając ośmiu pierwiastkom w drugim okresie, od litu do neonu. Dla n = 3 można oczekiwać, że w tym okresie będzie 18 pierwiastków, ale dzieje się tak, że z 18 możliwych elektronów dodatkowych jest tylko osiem, które zajmują zewnętrzną powłokę argonu, który kończy trzeci okres obejmujący 8 pierwiastków. W przypadku potasu, następnego pierwiastka po argonie, nowa powłoka zaczyna się od elektronu o n = 4, a powłoka elektronowa o n = 3 zaczyna się dopiero teraz wypełniać. Powłoka n = 4 zawiera 8 elektronów wypełniających zewnętrzną powłokę kryptonu, a okres zawiera 18 pierwiastków.
W następnym okresie powłoka n = 5 zaczyna się zapełniać poczynając od rubidu i osiąga 8 elektronów dla ksenonu, dla którego powłoka n = 4 osiągnęła 18 elektronów. Następny okres rozpoczynający się od cezu zapełnia elektronami zewnętrzną powłokę n = 6; od lantanu powłoka n = 4 zaczyna się wypełniać do lutetu gdzie osiąga swoje maksymalne wypełnienie 32 elektronami, przy czym zewnętrzne powłoki 1(n = 5) + 2 (n = 6) elektronów walencyjnych pozostają niezmienione. Zatem 15 pierwiastków ziem rzadkich od lantanu do lutetu ma bardzo podobne właściwości. Od hafnu do radonu powłoka n = 5 zostaje zapełniona do 18, a powłoka n = 6 do 8 elektronów. Ten okres, od cezu do radonu, zawiera 32 pierwiastki. Następny okres, zaczynający się od pierwiastka fransu, zawiera elektrony na powłoce n = 7. Jest niekompletny, ostatnim naturalnym pierwiastkiem jest uran (liczba atomowa 92), a wszystkie zawarte w nim pierwiastki są radioaktywne. Niektóre wyższe pierwiastki w tym okresie uzyskano sztucznie.
Elektronowa teoria wartościowości
W poprzedniej teorii wartościowości nie rozróżniono związków jonowych, takich jak chlorek sodu, i substancji niejonowych, takich jak dwutlenek węgla. Teorie zaproponowane przez A. Kossela i G. N. Lewisa uznają, że są one zasadniczo różne. Jeśli zewnętrzne elektrony w atomie są oznaczone kropkami, tworzenie chlorku sodu i cząsteczki chloru może być przedstawione jako:
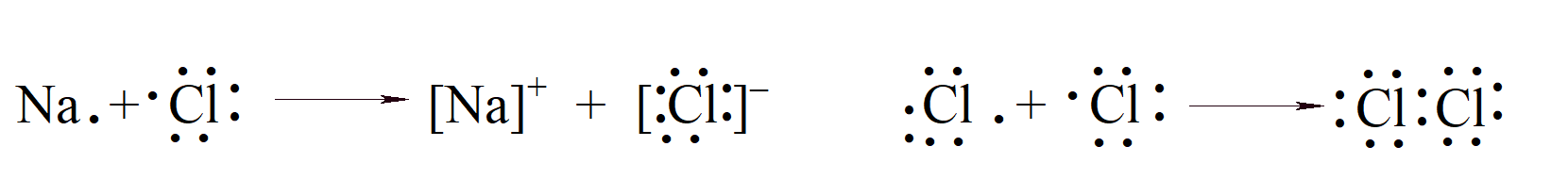
NaCl zawiera obdarzone ładunkiem jony (już obecne w krysztale) utrzymywane razem siłami elektrostatycznymi bez rzeczywistego wiązania walencyjnego; druga cząsteczka jest obojętna, w której składowe są utrzymywane razem dzięki wiązaniu walencyjnemu lub kowalencyjnemu, tworzonemu przez wspólną parę elektronów o przeciwnych spinach. Podwójne wiązanie tworzą dwie wspólne pary elektronów, a potrójne wiązanie - trzy wspólne pary, czemu odpowiadają kreski obrazujące wiązania we wzorach cząsteczek np. O=C=O, N≡N. W niektórych przypadkach cząsteczka zawiera nieparzysty elektron jak w przypadku tlenku azotu(II) O=N· czy dwutlenku chloru ·ClO2, takie cząsteczki są paramagnetyczne.
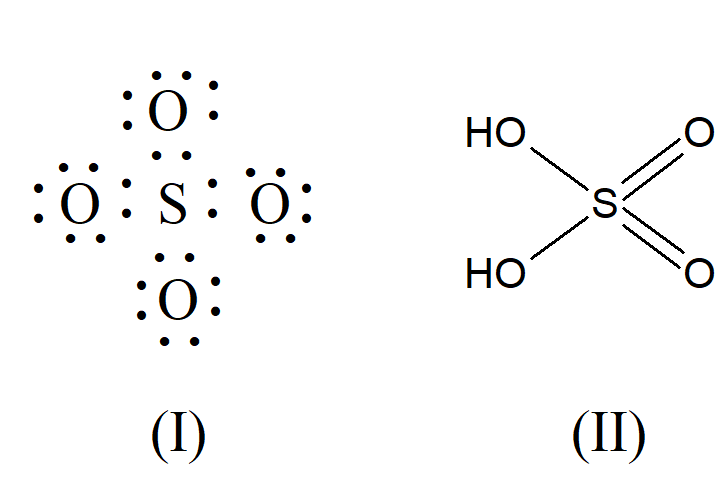
Przy tworzeniu struktur jonów kwasów tlenowych I. Langmuir (1919 r.) zastosował inny rodzaj wiązania, który G. A. Perkins nazwał „wiązaniem zapożyczonym”. Jest to wiązanie kowalencyjne, w którym oba elektrony pary pochodzą od jednego atomu zamiast po jednym elektronie z każdego z dwóch atomów. Langmuir reprezentował jon siarczanowy(VI) SO42– jako utworzony przez atom siarki z sześcioma elektronami walencyjnymi do których dołączyły dwa kolejne przekształcając go w jon S2–. Cztery pary elektronów tego jonu tworzą wiązania z czterema atomami tlenu, przy czym wszystkie atomy mają oktety elektronowe jak to pokazano w strukturze (I), podczas gdy struktura oparta na konwencjonalnej wartościowości ma dwa wiązania pojedyncze i dwa wiązania podwójne, jak w kwasie siarkowym(VI) (II):
Przypadek azotu jest interesujący. Chociaż na zewnętrznej powłoce atomu jest pięć elektronów, nie tworzą one pięciu wiązań, a azot nigdy nie ma wartościowości równej pięć, jak wcześniej zakładano, że ma to miejsce w przypadku związków amonowych, takich jak NH4Cl i kwas azotowy HO·NO2 Wszystkie związki amonowe zawierają jon NH4+, który powstaje przez przyłączenie protonu H+ (jądra atomu wodoru) do wolnej pary elektronów na atomie azotu w cząsteczce amoniaku, przy czym maksymalna wartościowość azotu wynosi cztery:
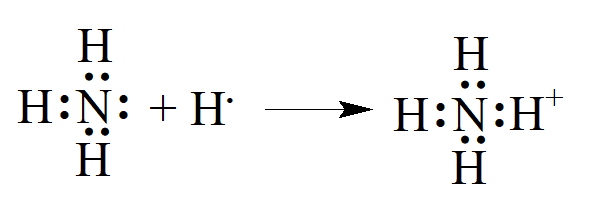
Do jonu azotanowego(V) dodaje się dodatkowy elektron, zwiększając ich liczbę na atomie azotu do sześciu i nadając jonowi ładunek ujemny.
Kwas azotowy(V) zachowuje się w wielu reakcjach ze związkami organicznymi, jakby miał strukturę HO·NO2, np. w nitrowaniu benzenu:
C6H6 + HO·NO2 → C6H6NO2 + H2O.
Grupa nitrowa NO2 została pierwotnie określona jako zawierająca pięciowartościowy azot, a kwas azotowy(V) w formie (a). Kwas azotowy(V) można przedstawić jako zawierający czterowartościowy azot (jak w jonie amonowym) poprzez zastąpienie jednego podwójnego wiązania między azotem i tlenem pojedynczym wiązaniem utworzonym przez parę elektronową zlokalizowaną na azocie przenoszoną do atomu tlenu, jak pokazano na (b) [należy pamiętać, że to historia chemii!]:
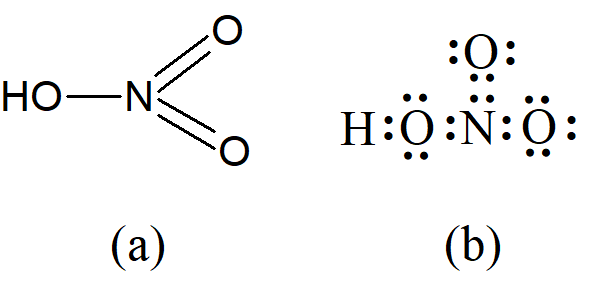
M. Lowry i N. Sidgwick traktowali takie wiązania utworzone przez oddanie pary elektronów z jednego atomu zamiast dzielenia elektronu z każdego z dwóch atomów, inaczej niż zwykłe pojedyncze wiązania. Ponieważ oddanie pary elektronów przez jeden atom nada mu ładunek dodatni, a atom który przyjmuje tę parę uzyskuje ładunek ujemny, Lowry uznał takie wiązanie za połączenie elektrowalencyjności i kowalencyjności, a zatem formalnie jako wiązanie podwójne, które nazwał „mieszanym podwójnym wiązaniem”, reprezentowanym przez A+—B–. Sidgwick, który zauważył, że takie wiązania są obecne w związkach koordynacyjnych, nazwał to wiązanie „wiązaniem koordynacyjnym” i przedstawiał je strzałką skierowaną do atomu, który uzyskiwał ładunek ujemny, A→B. Sidgwick sformułował również zasadę, że maksymalne kowalencyjności w drugim okresie układu okresowego wynoszą 4, tak że azot w kwasie azotowym(V) ma cztery wiązania, a kwas siarkowy(VI) można zapisać w podobny sposób, z dwoma wiązaniami koordynacyjnymi na atomie siarki:
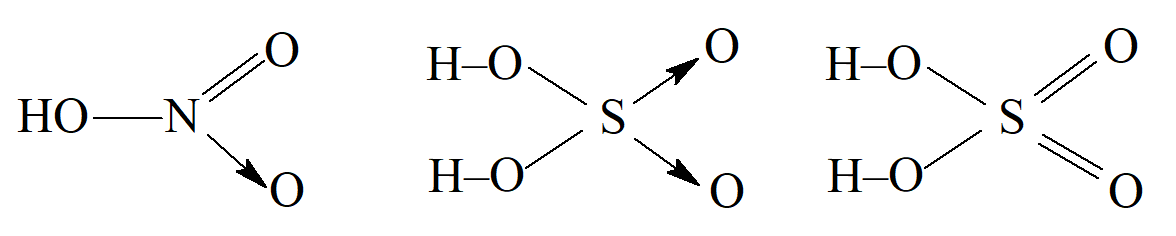
Istnieje wiele powodów, dla których można przypuszczać, że wiązania między azotem a tlenem w kwasie azotowym(V) są takie same, podczas gdy wzór powyższy sugeruje, że w grę wchodzą trzy różne rodzaje wiązań. J. Pauling zasugerował, że rozkład elektronów jest ujednolicony przez szybkie zmiany położenia par elektronów do różnych możliwych konfiguracji w cząsteczce, i nazwał ten proces „rezonansem”, dzięki czemu wszystkie wiązania są identyczne. W przypadku cząsteczek zawierających nieparzysty elektron nie uważa się go za umiejscowiony na jednym konkretnym atomie, ale za współdzielony, w wyniku rezonansu, między różnymi atomami w cząsteczce.
Nie należy przypuszczać, że związek wykazujący rezonans jest mieszaniną cząsteczek o różnych konfiguracjach, ale że wszystkie cząsteczki są identyczne i zawierają wkłady różnych możliwych struktur rezonansowych. Teorię tę zastosowano do benzenu, który wzór Kekulé przedstawia jako zawierający naprzemienne wiązania pojedyncze i podwójne w pierścieniu. Dzięki rezonansowi wszystkie wiązania są takie same i mają charakter pośredni między wiązaniami pojedynczymi i podwójnymi.
Atomy cięższe od helu mają więcej niż jedną powłokę elektronową, Bohr próbował określić układ elektronów na kolejnych powłokach na podstawie widm atomowych. Jego schemat został zmodyfikowany przez C. R. Bury’ego (1921 r.) i Main Smitha (1924 r.). W niektórych przypadkach elektrony powłoki leżącej poniżej zewnętrznej mogą funkcjonować jako elektrony walencyjne przez co można wyjaśnić zmienne wartościowości niektórych pierwiastków, szczególnie pierwiastków przejściowych mających niepełne powłoki wewnętrzne. W przypadku pierwiastków ziem rzadkich elektrony dodawane do atomu wraz ze wzrostem ładunku jądrowego zajmują powłokę znajdującą się wewnątrz struktury elektronowej, dzięki czemu ich wartościowość nie ulega zmianie, inaczej niż w przypadku większości pierwiastków.
Związki koordynacyjne
Na wczesnym okresie teorii wartościowości odczuwano wiele trudności w wyjaśnianiu tego, co często nazywano „związkami cząsteczkowymi” czyli takimi, które powstają przez łączenie cząsteczek, w których wszystkie wartościowości są wysycone. Wiele z nich jest bardzo trwałych. Chlorek platyny PtCl4 łatwo łączy się z kwasem chlorowodorowym z wytworzeniem H2PtCl6, i stwierdzono, że zawiera on rodnik PtCl6, który występuje również w wielu solach, takich jak K2PtCl6. Wartościowość platyny przyjęto jako równą dwa w związkach takich jak PtCl2, i cztery w związkach takich jak PtCl4. W H2PtCl6 jest ona najwyraźniej równa sześć. Wiele soli łączy się z amoniakiem, tworząc związki zwane amoniakatami, i niektóre z nich są bardzo stabilne, w szczególności związki zawierające kobalt(II), których proste związki raczej łatwo ulegają utlenieniu. Chociaż CoCl3 nie jest znany jako taki, znanych jest wiele zawierających go związków, szczególnie w połączeniu z amoniakiem, takich jak CoCl3·6NH3. Podjęto próby wyjaśnienia zakładając, że azot jest pięciowartościowy, np. Ch. W. Blomstand (1869 r.), ale pierwszą udaną teorię zaproponował Alfred Werner (1866–1919) w 1893 r.
Pierwszym literaturowym doniesieniem o syntezie związku koordynacyjnego był artykuł francuskiego chemika B. M. Tassaerta z roku 1798. Opisał on reakcję pomiędzy chlorkiem kobaltu(III) a roztworem amoniaku w wyniku czego uzyskał różowy osad, którego skład został określony jako CoCl3.6NH3. Wcześniej oczywiście znane były inne związki koordynacyjne, jak przykładowo błękity Pruski czy Turnbula, lecz nie publikowano doniesień na ich temat. W następnych latach po publikacji Tassaerta otrzymano szereg związków kobaltu(III) czy platyny(II) z amoniakiem, ale budowa cząsteczek tych związków stanowiła nierozstrzygnięty problem. Szczególne znaczenie uzyskały związki kobaltu(III), ponieważ ich analiza pozwoliła na wysunięcie dwóch teorii budowy tego typu połączeń chemicznych.
|
wzór |
kolor |
Ilość jonów Cl– strąconych z roztworu związku za pomocą Ag+ (Blomstrand) |
przewodnictwo (Werner) |
|
CoCl3(NH3)6 |
żółty |
3 |
wysokie |
|
CoCl3(NH3)5 |
purpurowy |
2 |
średnie |
|
CoCl3(NH3)4 |
zielony (trans) |
1 |
niskie |
|
CoCl3(NH3)4 |
fioletowy (cis) |
1 |
niskie |
|
CoCl3(NH3)3 |
pomarańczowy |
0 |
brak przewodnictwa |
W drugiej połowie XIX wieku szwedzki chemik Blomstrand i niezależnie od niego S. M. Jørgensen bazując na analizie ilości jonów chlorkowych jakie były strącane z roztworów związków kobaltu(III) z amoniakiem zaproponowali łańcuchową budowę tego typu połączeń. Zgodnie z tym strukturę tych związków można było przedstawić następująco:
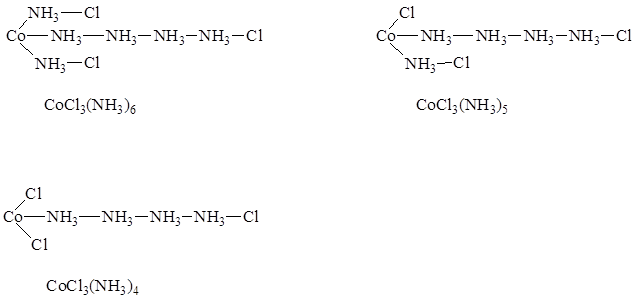
Jak łatwo zauważyć układ łańcuchowy cząsteczek amoniaku przypomina łańcuchy węglowe charakterystyczne dla związków organicznych. Takie podejście wyjaśniało różnice w ilości jonów chlorkowych strącanych jonami srebra z roztworów tych związków.
Druga koncepcja budowy tego typu związków pojawiła się w roku 1893 za sprawą Alfreda Wernera. Ten niemiecki chemik oparł swoje rozważania o pomiary konduktometryczne roztworów tych samych związków kobaltu. Na podstawie swoich badań zaproponował teorię wartościowości wyjaśniającą budowę związków koordynacyjnych. Zgodnie z jego koncepcją pierwiastki wykazują dwa rodzaje wartościowości: główną i poboczną dążąc do wysycenia obydwu. Wartościowość poboczna dodatkowo jest skierowana w konkretne miejsca przestrzeni. Zgodnie z jego koncepcją przedstawione powyżej związki można opisać wzorem [Co(NH3)6-nCln]X3-n, gdzie n <0,1,2>, a ich rzeczywista budowa jest następująca:
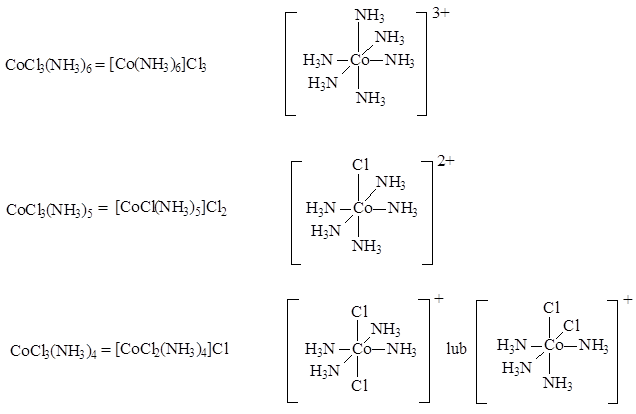
Werner wprowadził pojęcie związku koordynacyjnego oraz pojęcie liczby koordynacyjnej. Zaproponowana przez niego struktura oktaedryczna dla związków koordynacyjnych pozwoliła na wyjaśnienie ilości jonów chlorkowych strącanych jonami srebra, analogicznie jak teoria Blomstranda, a ponadto pozwalała na wyjaśnienie danych konduktometrycznych. Dodatkowo za oktaedrycznym otoczeniem jonu kobaltu(III) wyjaśnia istnienie tylko dwóch izomerów [CoCl2(NH3)4]Cl. Ostatecznym potwierdzeniem słuszności teorii Wernera była synteza analogicznego związku kobaltu z ligandem dietylenoaminowym, którego izomer cis wykazuje izomerię optyczną w odróżnieniu od izomeru trans.
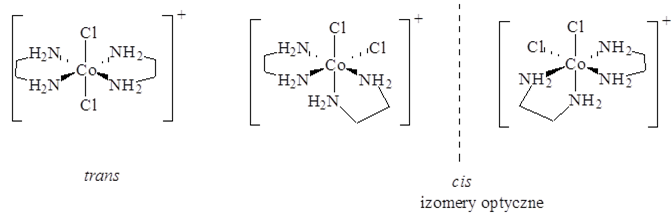
Problemem jaki napotkała teoria Wernera jest reguła oktetu, którego rozwiązaniem było uwzględnienie orbitali d jonu centralnego. Jednak na takie rozwiązanie problemu należało poczekać do opracowania budowy elektronowej pierwiastków i związków chemicznych.
Biorąc pod uwagę teorię Wernera możemy zdefiniować związki koordynacyjne jako związki chemiczne, w których można wyróżnić jeden lub więcej atomów (jonów) centralnych otoczonych przez inne atomy (jony) lub cząsteczki zwane ligandami, przy czym przynajmniej jedno wiązanie chemiczne w tym układzie musi mieć charakter wiązania koordynacyjnego. Liczbę koordynacyjną określa się jako sumaryczną liczbę wiązań koordynacyjnych σ pomiędzy atomem (jonem) centralnym a ligandami. Cechą charakteryzującą atom (jon) centralny związku koordynacyjnego jest jego stopień utlenienia, który jest formalnym ładunkiem przypisywanym temu atomowi (jonowi). Stopień utlenienia nie jest równoznaczny z wartościowością atomu (jonu) centralnego, ani z rzeczywistym ładunkiem na atomie (jonie) centralnym.
Ze względu na całkowity ładunek cząsteczki związku koordynacyjnego możemy podzielić kompleksy na:
a) jonowe (kationowe, anionowe)
b) obojętne
Ze względu na liczbę atomów centralnych w cząsteczce związku koordynacyjnego dzielimy je na:
a) związki koordynacyjne monojądrowe – jeden atom centralny
b) związki koordynacyjne wielojądrowe – dwa lub więcej atomów centralnych
c) klastery – trzy lub więcej atomów (jonów) centralnych, miedzy którymi występują wiązania
Ligandy w związkach koordynacyjnych mogą być pojedynczymi jonami pierwiastków jak przykładowo jony chlorowców, atomami (np. tlen) lub cząsteczkami nieorganicznymi (np. N2, NO, CO, H2O) i organicznymi. Jeżeli w związku koordynacyjnym występuje wiązanie metal–węgiel to związki tego rodzaju nazywane są związkami metaloorganicznymi. Ligandy ze względu na sposób koordynacji do atomu (jonu) centralnego dzieli się na:
a) jednodonorowe – jeden atom donorowy w cząsteczce liganda
b) wielodonorowe – bi–, tri–, tetra–, itd. donorowe, w których występują odpowiednio 2, 3, 4 atomy donorowe wiążące się z jonem centralnym.
Inny podział ligandów oparty jest na ich właściwościach donorowo–akceptorowych. Z tego względu wyróżnia się ligandy σ–donorowe, π–donorowe i π–akceptorowe.
Dalsze losy historii praw, idei i koncepcji chemii można znaleźć na stronach serwisu.