 Principles of Chemistry
Principles of Chemistry
 Odpychanie par elektronowych na powłoce walencyjnej
Odpychanie par elektronowych na powłoce walencyjnej
Koncepcja odpychania par elektronowych na powłoce walencyjnej w skrócie VSEPR od anielskiej nazwy Valence Shell Electron Pair Repulsion, nie jest modelem wiązania chemicznego, a pewną koncepcją pozwalającą na przewidywanie budowy geometrycznej cząsteczek. Koncepcja opiera się na teorii wiązań walencyjnych i pewnych danych eksperymentalnych. Powstanie tego modelu zawdzięczamy Sidgwickowi i Powellowi, którzy zauważyli, że budowę geometryczna wielu prostych cząsteczek o wiązaniu kowalencyjnym można wyjaśnić przyjmując, że pary elektronowe atomu centralnego wzajemnie się odpychają. Tetraedryczna geoemtria cząsteczki metanu wynika, w związku z tym, z odpychania czterech par elektronowych tworzących cztery wiązania kowalencyjne pomiędzy węglem i wodorem. Podobnie w przypadku amoniaku występują cztery pary elektronowe, ale ze względu na to, że jeden z wierzchołków czworościanu zajmuje wolna (nie tworząca wiązania) para elektronowa, cząsteczka amoniaku ma kształt piramidy.
Ogólna zasada przewidywania przestrzennego ułożenia wokół atomu polega na obliczeniu liczby elektronów (par elektronowych) w jego powłoce walencyjnej, a następnie na rozlokowaniu ich w przestrzeni w możliwie największej odległości od siebie. Zakłada się, że tylko pojedyncze pary tworzą wiązania chemiczne. Zgodnie z tym podejściem spodziewać się należy, że atomy o takiej samej liczbie elektronów będą miały takie samo przestrzenne rozmieszczenie par elektronowych.
| liczba par elektronowych |
kształt | przykład | |
| 2 | linia | |
MgF2 |
| 3 | trójkąt | BF3 | |
| 4 | tetraedr |  |
CF4 |
| 5 | podwójna piramida trygonalna |
 |
PF5 |
| piramida tetragonalna |  |
[InCl5]2– | |
| 6 | oktaedr |  |
SF6 |
| 7 | podwójna piramida pentagonalna |
 |
IF7 |
Stosunkowo łatwo jest przewidzieć konfigurację związku chemicznego posługując się koncepcją VSEPR. Konfiguracje dla pierwiastków s i p-elektronowych przewidywane na gruncie tej koncepcji i wyznaczone eksperymentalnie są zgodne. jest to niewątpliwa zaleta tej teorii jednak znacznie trudniej jest znaleźć jej teoretyczne uzasadnienie. Najczęściej wyjaśnienia teoretyczne opiera się na regule (zakazie) Pauliego. Zgodnie z tym zakazem dwa elektrony o takich samych spinach unikają się wzajemnie, a tym samym jeżeli powłoka walencyjna zawiera 2n elektronów to n z nich zajmie pozycje jak najbardziej oddalone od siebie, czyli w wierzchołkach odpowiedniego wielościanu. Ligandy zbliżające się do takiego atomu będą mogły uwspólniać elektrony (tworzyć wiązania) zbliżając się do tych wierzchołków wielościanu, w których znajdują się elektrony atomu centralnego. Jednocześnie pozostałe n elektronów o przeciwnym spinie będzie również skierowane w stronę zbliżających się ligandów. W ten sposób elektrony będą ułożone parami w stronę wierzchołków wielościanu i w ten sposób powstają konfiguracje cząsteczek przytoczone powyżej. Wadą tego wyjaśnienia jest uznanie, że korelacja spinów elektronów odgrywa decydującą rolę w tworzeniu struktury cząsteczki.
Pierwotna koncepcja Sidgwicka i Powella została rozszerzona przez Nyholma i Gillespiego, którzy uwzględnili fakt, że wolne pary elektronowe różnią się od wiążących par elektronowych. Wolne pary elektronowe są zlokalizowane na atomie centralnym i nie są dzielone z innymi atomami. W związku z tym ich wzajemne oddziaływanie jest silniejsze niż odpychanie dwóch wiążących par elektronowych. Z tego wynika kolejność siły oddziaływań:
wolna para–wolna para > wolna para–wiążąca para > wiążąca para–wiążąca para.
Weźmy pod uwagę cząsteczkę SF4. Możemy się dla niej spodziewać, zgodnie z powyższymi rysunkami struktury podwójnej piramidy trygonalnej, lub piramidy o podstawie kwadratowej, gdyż cząsteczka ta na powłoce walencyjnej ma 10 elektronów (6 od siarki i po jednym od atomów fluoru). mamy pięć par elektronowych i cztery atomy fluoru związane z siarką, czyli pozostaje jedna niewiążąca (wolna) para elektronowa. Jeżeli wolna par elektronowa zajmie jedną z pozycji na osi podwójnej piramidy trygonalnej to nastąpią trzy oddziaływania typu odpychania pomiędzy parą wiążącą.
| a) | b) |
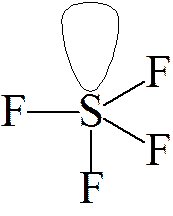 |
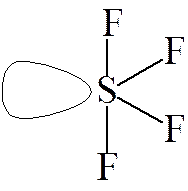 |
Jeżeli jednak wolna para znajdzie się w którymś położeniu horyzontalnym, jak na rysunku b), to wystąpią tylko dwa oddziaływania odpychające, co jest bardziej korzystne. Uwzględniając różnice w oddziaływaniach pomiędzy wolnymi parami elektronowymi między soba i z parami wiążącymi należy spodziewać się, że w przypadku cząsteczki SF4 kat pomiędzy atomami fluoru znajdującymi się na osi cząsteczki będzie mniejszy od 180°. Rzeczywiście kąt ten wynosi 178° co wynika z różnicy w sile oddziaływań pomiędzy wolna parą elektronowa a parą wiążącą.
Zgodnie z koncepcją VSER wiązanie podwójne zajmuje tylko jedno miejsce koordynacji ale tworzą je cztery elektrony. Z tego względu odpychanie w stosunku do innych par elektronowych jest większe. Rozpatrując kąty w cząsteczkach SF3 i amoniaku, daje się zauważyć, że zmiana wodoru na fluor powoduje zmiany kątów. W amoniaku kąt H–N–H wynosi 106,6°, a w przypadku fluorku azotu kąt F–N–F wynosi tylko 102,1°. Fakt ten tłumaczy się większą elektroujemnością fluoru, który silnie przyciąga ładunek i przez to zmniejsza siłę oddziaływania pomiędzy wiążącymi parami elektronowymi związanymi z atomem azotu. Analogiczne rozumowanie dotyczy zmiany kątów H–A–H w szeregu:
| NH3 | PH3 | AsH3 | SbH3 |
| 106,6° | 93,9° | 91,8° | 91,3° |
w którym wzrastające donorowanie ładunku od cięższych pierwiastków do wodoru powoduje zmniejszenie siły odpychania pomiędzy wiążącymi parami elektronowymi.
Krytyka koncepcji VSEPR
Koncepcja ta jest bardzo skuteczna w odniesieniu do związków pierwiastków p-elektronowych. Szczególnie jest to widoczne gdy uświadomimy sobie jak niewielkie różnice energetyczne występują pomiędzy różnymi dopuszczalnymi strukturami cząsteczek związków. Dla amoniaku energia inwersji czyli przejścia atomu azotu przez płaszczyznę wyznaczoną atomami wodoru wynosi jedynie 25 kJ/mol, co wskazuje na to, że płaska struktura cząsteczki jest niewiele mniej trwała od tetraedrycznej. Z drugiej strony niepowodzenia teorii VSEPR wynikają z równie niewiele znaczących efektów fizycznych. Największym problemem jaki napotyka ta koncepcja jest trudność w zdefiniowaniu powłoki walencyjnej. Weźmy pod uwagę anion [TeCl6]3–, który będzie miał strukturę oktaedryczną. Jednak w soli amonowej (NH4)3[TeCl6] liczba 14 elektronów wskazuje na strukturę zdeformowaną. Podobnie anion [SbBr6]3– o 14 elektronach walencyjnych i geometrii oktaedrycznej w roztworze wykazuje strukturę zdeformowaną. W przypadku jonu telluru możemy założyć, że orbitals 5s jest chemicznie obojętny i układ ma 12 elektronów walencyjnych i strukturę oktaedrtyczną. Problem zdefiniowania powłoki walencyjnej jest szczególnie istotny dla pierwiastków przejściowych oraz f-elektronowych. W tych przypadkach koncepcja VSEPR jest całkowicie nieprzydatna, ponieważ dla związków tych metali nie można wyróżnić zlokalizowanych wiązań kowalencyjnych.
Zmniejszenie udziału orbitali s w wiązaniu może tłumaczyć zmianę kątów pomiędzy wiązaniami w szeregu wodorków AH3. Jeżeli orbitale p antymonu biorą udział w wiązaniu SbH3, to można się spodziewać, że kąty będą wynosiły 90° co jest wartością zbliżoną do wyznaczonej eksperymentalnie. Stopniowe zwiększanie kąta miedzy wiązaniami w miarę przechodzenia od SbH3 do amoniaku może wskazywać na wzrost udziału orbitali s w wiązaniu. Na tej podstawie można wskazywać, że wolna para elektronowa azotu będzie miała bardziej charakter zhybrydyzowanego orbitalu sp3 niż bezkierunkowego orbitalu s. To z kolei wskazuje na bardziej zasadowe właściwości amoniaku, co jest zgodne z rzeczywistością. Udział orbitali s można, zgodnie ze spostrzeżeniem Pearsona, opisywać za pomocą efektu Jahna-Tellera drugiego rządu, czyli odkształcenia struktury związku wywołuje mieszanie orbitali s i p atomu centralnego. Ten proces prowadzi do stabilizacji cząsteczki.
Koncepcja VSEPR całkowicie zawodzi jeżeli w cząsteczce występują wiązania zdelokalizowane. Przykładowo cząsteczki C(CN)3–, C(NO2)3– i (SiH3)3N są izoelektronowe, mają po 8 elektronów walencyjnych, to mają struktury płaskich trójkątów, co wynika z obecności zdelokalizowanych orbitali π. Podobna sytuacja ma miejsce w przypadku mostkujących atomów tlenu i fluoru wykazujących szeroki zakres kątów miedzy wiązaniami. Spodziewana struktura tetraedryczna, dla czterech par elektronowych, występuje w tych układach niezwykle rzadko, co jest najprawdopodobniej wynikiem obecności zdelokalizowanych, trójcentrowych wiązań jakie występują w tych układach mostkowych.
Użyteczną cechą koncepcji VSEPR jest to, że na podstawie przewidywanego rozkładu przestrzennego par elektronowych można przewidywać reaktywność związków. Oczywistym jest, że związek o strukturze, w której występuje ukierunkowana, wolna para elektronowa, może zachowywać się jak zasada Lewisa. W przypadku odwrotnym gdy cząsteczka nie posiada wolnych par elektronowych i wykazuje "otwartą" strukturę np. trójkątną, może przyjmować parę elektronową, zachowując się jak kwas Lewisa. Typowymi przykładami zasad Lewisa są SnCl3–, P(CH3)3, SO32– o geometrii tetraedrycznej, a typowymi kwasami Lewisa BF3, zmiana geometrii z trójkątnej do tetraedrycznej, SbF5– od podwójnej piramidy trygonalnej do oktaedru.
Koncepcja VSEPR pozwala na przewidywanie, w prosty sposób, budowy geometrycznej dużej liczby związków pierwiastków p-elektronowych, dodatkowo pozwalając w stosunkowo prosty sposób przewidzieć kiedy przewidywana budowa związku na jej podstawie zawiedzie. Przewidywanie struktury związku na podstawie teorii orbitali cząsteczkowych jest trudniejsze, ale ze względu na większą liczbę uzyskiwanych parametrów bardziej wiarygodne.