 Principles of Chemistry
Principles of Chemistry
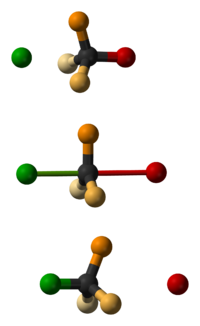 Rodzaje reakcji chemicznych
Rodzaje reakcji chemicznych
Niemalże wszystkie reakcje chemiczne składają się z szeregu prostych etapów, w których powstają i ulegają dalszym przemianom produkty pośrednie. Pojedyncze etapy można powiązać ze strukturą cząsteczek związków biorących udział w reakcjach, i ponadto dokonać klasyfikacji elementarnych etapów reakcji.
Generalnie atomy w cząsteczkach mogą ulegać dwóm typom przemian chemicznych, odpowiednio zmianom w sferze koordynacyjnej lub też zmianie stopnia utlenienia. Trzecia możliwość związana jest z reakcjami pomiędzy ligandami występującymi w sferze koordynacyjnej danego atomu centralnego. Ten trzeci rodzaj jest istotny dla związków koordynacyjnych, ale obejmuje również procesy, w których nie uczestniczy atom centralny, a centra reakcji znajdują się w cząsteczkach ligandów połączonych z danym atomem. Pewną niedogodnością takie podziału jest to, że wiele reakcji chemicznych można zaliczyć do więcej niż jednego rodzaju. Jest to wynikiem pewnej sztuczności zastosowanej klasyfikacji, która poza tym jest bardzo dogodna.
Opierając się na przedstawionej klasyfikacji można reakcje chemiczne podzielić na:
- Zmiana w sferze koordynacyjnej
- reakcje przyłączania – wzrost liczby koordynacyjnej
- reakcje eliminacji – zmniejszenie liczby koordynacyjnej
- reakcje podstawienia (substytucji) – wymiana ligandu
- zmiana geometrii – zmiany kątów wiązania metal–ligand bez zrywania wiązania
- zmiana rozmieszczenia ligandów – przykładowo izomeryzacja cis – trans
- reakcje pomiędzy ligandami prowadzące do zmian w sferze koordynacyjnej
- Reakcje przeniesienia elektronów
- utrata lub przyjęcie elektronów bez zmian w sferze koordynacyjnej
- reakcje redoks połączone ze zmianami w sferze koordynacyjnej
Reakcje przyłączenia i eliminacji
MLn + L → MLn+1
MLn – L → MLn–1 + L
Opis tych reakcji można oprzeć na mechanizmach dotyczących procesów homo– i heterolitycznych. Reakcje elektrofilowe i nukleofilowe różnią się mię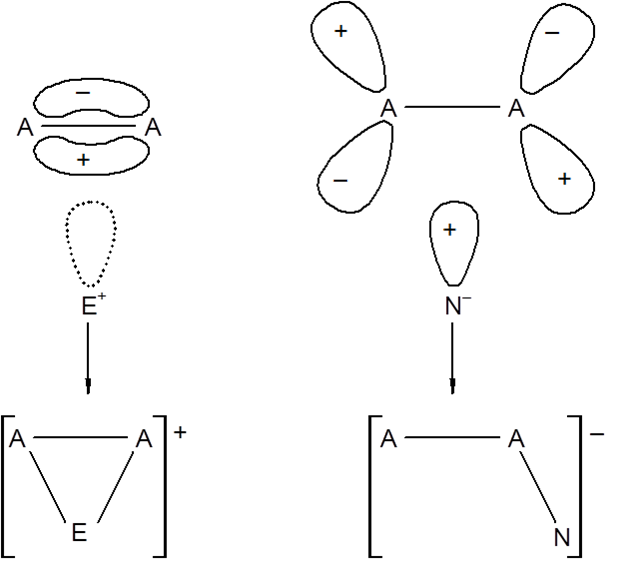 dzy sobą jedynie tym, który ze związków pełni rolę substratu. Jeden atom musi mieć obsadzony, parą elektronową, orbital, a drugi pusty orbital akceptorowy. Na ogół przyłączanie ligandów prowadzące do powiększeniem liczby koordynacyjnej łączy się ze zmianą położeń ligandów obecnych w sferze koordynacyjnej. Orbitale akceptorowe lub donorowe mogą być orbitalami cząsteczkowymi, i w zależności od rodzaju tych orbitali można oczekiwać różnych produktów reakcji.
dzy sobą jedynie tym, który ze związków pełni rolę substratu. Jeden atom musi mieć obsadzony, parą elektronową, orbital, a drugi pusty orbital akceptorowy. Na ogół przyłączanie ligandów prowadzące do powiększeniem liczby koordynacyjnej łączy się ze zmianą położeń ligandów obecnych w sferze koordynacyjnej. Orbitale akceptorowe lub donorowe mogą być orbitalami cząsteczkowymi, i w zależności od rodzaju tych orbitali można oczekiwać różnych produktów reakcji.
Na rysunku obok przedstawiono oddziaływanie układu elektrofilowego z orbitalami π i układu nukleofilowego z orbitalem π*. Produkty obydwu reakcji sa różne co wynika z różnic w budowie przestrzennej orbitali wiążących i antywiążących.
Reakcje eliminacji zachodzą łatwiej gdy sfera koordynacyjna jest zniekształcona i ściśnięta. Heterolityczna eliminacja zachodzi łatwiej gdy rozpuszczalnik polarny, w którym prowadzona jest reakcja stabilizuje powstające kwasy lub zasady Lewisa. Dysocjacja czyli homolityczne usunięcie substancji nie będącej rodnikiem zachodzi stosunkowo trudno i wymaga aktywacji termicznej lub fotochemicznej. Stosunkowo łatwo natomiast zachodzą reakcje eliminacji rodników z większych cząsteczek będących również rodnikami. Tego typu procesy są często skojarzone z przeniesieniem elektronów.
Reakcje podstawienia
MLn + L’ → MLn–1L’ + L
W każdej tego typu reakcji musi nastąpić zerwanie jednego wiązania i powstanie nowego. Reakcje te przebiegają według dwóch głównych mechanizmów. W mechanizmie asocjacyjnym następuje wstępne przyłączenie cząsteczki liganda L’, a następnie odszczepienie cząsteczki liganda ustępującego. W mechanizmie dysocjacyjnym pierwszym etapem jest usunięcie liganda L ze sfery koordynacyjnej a następnie przyłączenie liganda L’. W pierwszym przypadku w etapie przejściowym mamy do czynienia ze związkiem o zwiększonej liczbie koordynacyjnej, a w drugim związek przejściowy jest koordynacyjnie niewysycony. Możliwy jest też mechanizm wzajemnej wymiany, w którym przerywanie i tworzenie wiązania zachodzi równolegle, a obydwa etapy wpływają na siebie wzajemnie. W mechanizmie wzajemnej wymiany wyróżnia się dwa rodzaje. W pierwszym z nich zbliżenie liganda wchodzącego do sfery koordynacyjnej jest poprzedzone nieznacznym osłabieniem wiązania M–L. Natomiast w drugim osłabienie wiązania M–L poprzedza zbliżenie liganda L’. Mechanizmy te oznacza się symbolami klas A – dla mechanizmu asocjacyjnego, D – dla dysocjacyjnego oraz I dla mechanizmu wzajemnej wymiany. Ostatnia klasa została podzielona na dwie podklasy Ia oraz Id. W mechanizmie Ia charakter liganda L’ wykazuje wpływ na energię aktywacji i szybkość procesu wymiany, a w mechanizmie Id takiego wpływu się nie obserwuje.
W wyjaśnieniu mechanizmów reakcji dużą role odgrywa stereochemia produktów. W przypadku mechanizmu asocjacyjnego konfiguracja związku o geometrii tetraedrycznej ulega odwróceniu w wyniku podstawienia liganda.
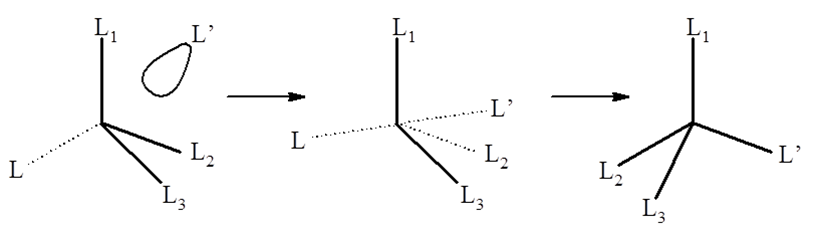 W mechanizmie wzajemnej wymiany typu Ia jako pośredni występuje związek o geometrii trójkątnej, do którego nad lub pod płaszczyzną zbliża się ligand L’. W mechanizmie Ia zakłada się maksymalne nakrywanie orbitali cząsteczki L’ z antywiążącym orbitalem M–L. W chemii nieorganicznej mechanizmy reakcji są bardziej skomplikowane niż ma to miejsce w chemii związków organicznych gdyż często mamy do czynienia z reakcjami następczymi, a zwiększone liczby koordynacyjne produktów pośrednich są na porządku dziennym.
W mechanizmie wzajemnej wymiany typu Ia jako pośredni występuje związek o geometrii trójkątnej, do którego nad lub pod płaszczyzną zbliża się ligand L’. W mechanizmie Ia zakłada się maksymalne nakrywanie orbitali cząsteczki L’ z antywiążącym orbitalem M–L. W chemii nieorganicznej mechanizmy reakcji są bardziej skomplikowane niż ma to miejsce w chemii związków organicznych gdyż często mamy do czynienia z reakcjami następczymi, a zwiększone liczby koordynacyjne produktów pośrednich są na porządku dziennym.
Reakcje podstawienia nukleofilowego, zachodzące według wszystkich wymienionych typów są bardzo rozpowszechnione w chemii nieorganicznej. Wyjątek stanowią atomy na niskich stopniach utlenienia tworzące wiązania o silnie kowalencyjnym charakterze. Rzadziej występują reakcje podstawienia elektrofilowego, chociaż wymiana protonu i reakcje jonów metali z ujemnie naładowanymi centrami ligandów można uważać za podstawienie elektrofilowe.
Ograniczając się do reakcji podstawienia nukleofilowego można wyróżnić pewne cechy reakcji związków nieorganicznych. W przypadku pierwiastków grup głównych reakcje z udziałem związków o tetraedrycznej geometrii sfery koordynacyjnej reakcje przebiegają według wszystkich typów mechanizmów. Natomiast obecność mniejszych atomów sprzyja mechanizmom dysocjacyjnym. Natomiast cięższe pierwiastki wykazują skłonność do reakcji asocjacyjnych. Podobnie jony pierwiastków przejściowych na wysokich stopniach utlenienia uczestniczą w reakcjach asocjacyjnych, podczas gdy tetraedryczne jony związków metali d 10 elektronowych na niskich stopniach utlenienia wchodzą w reakcje dysocjacyjne. Mechanizm addycyjny jest preferowany przez związki o geometrii kwadratowej i konfiguracji d 10 atomu centralnego. Mechanizm dysocjacyjny jest preferowany przez związki o oktaedrycznej geometrii sfery koordynacyjnej. Związki o małych liczbach koordynacyjnych uczestniczą w reakcjach o mechanizmie asocjacyjnym.
Zmiana geometrii cząsteczki
W przypadku tych rekcji mamy dwie możliwości. W jednej nie następuje zerwanie wiązań, a w drugiej taki proces ma miejsce. W pierwszym przypadku można wyróżnić zjawisko płynności związków (obserwowane w przypadku niektórych związków koordynacyjnych gdzie koordynacja ligandów organicznych odbywa się poprzez wiązania σ lub π, i w których obydwie formy zmieniają się w bardzo szybko), inwersję układów o geometrii piramidalnej oraz przejście od konfiguracji tetraedrycznej do kwadratowej. Ponieważ nie następuje zrywani i tworzenie wiązań reakcje tego typu można uważać za oscylacje cząsteczki. 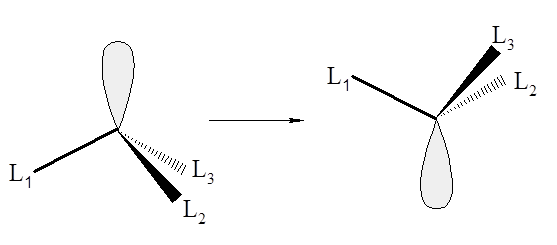 Zmiana konfiguracji związków koordynacyjnych Ni(II) z tetraedrycznej na kwadratową jest utrudniona ze względu na zmianę spinu aż o dwie jednostki. Dla układów piramidalnych o liczbie koordynacyjnej 3 w wyniku drgań oscylacyjnych zachodzi racemizacja, która jest bardzo szybka w przypadku amin, i znacznie wolniejsza dla fosfin. natomiast izomery soli tiolowe typu R1R2R3S+ udaje się rozdzielić metodami klasycznymi.
Zmiana konfiguracji związków koordynacyjnych Ni(II) z tetraedrycznej na kwadratową jest utrudniona ze względu na zmianę spinu aż o dwie jednostki. Dla układów piramidalnych o liczbie koordynacyjnej 3 w wyniku drgań oscylacyjnych zachodzi racemizacja, która jest bardzo szybka w przypadku amin, i znacznie wolniejsza dla fosfin. natomiast izomery soli tiolowe typu R1R2R3S+ udaje się rozdzielić metodami klasycznymi.
Izomeryzacja w przypadku związków koordynacyjnych metali przejściowych zachodzi najczęściej poprzez zerwanie wiązania metal–ligand, utworzenie związku o mniejszej liczbie koordynacyjnej a następnie utworzenie związku wysyconego koordynacyjnie o innym ułożeniu ligandów. Niemniej jednak w niektórych przypadkach proces racemizacji może przebiegać poprzez dystorsję geometrii sfery koordynacyjnej i w tym przypadku nie następuje zrywanie i tworzenie wiązań.