 Principles of Chemistry
Principles of Chemistry
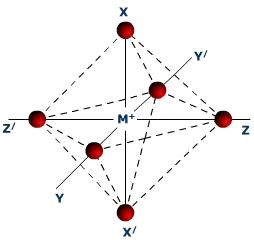 Cząsteczki związków metali przejściowych
Cząsteczki związków metali przejściowych
Nomenklatura związków koordynacyjnych została przedstawiona w dokumencie pdf do pobrania pod tym linkiem.
Opracowanie teorii pola krystalicznego i pola ligandów jest dostępne w postaci dokumentu pdf w tym miejscu.
Atomy pierwiastków przejściowych mają częściowo zapełnioną podpowłokę d przez co przez długi czas uważano, że opis wiązań w cząsteczkach ich związków wymaga innego podejścia niż stosowanego dla związków pierwiastków grup głównych. Jednak orbitale d atomu nie wykazują szczególnych właściwości kierunkowych, a obecność czterech płatów zwiększonej gęstości elektronowej w różnych kierunkach, jaka występuje w przypadku tych orbitali wskazuje na tendencję metali przejściowych do przyłączania większej liczby atomów. Innymi słowy pierwiastki te w związkach wykazują większe liczby koordynacyjne. Oczywiście pierwiastki f–elektronowe wykazują jeszcze większe liczby koordynacyjne, chociaż związek pomiędzy zapełnioną podpowłoką f a liczbą koordynacyjną jest niejednoznaczny.
Związki o dużych liczba koordynacyjnych, 4, 6 lub większych, zostały nazwane związkami koordynacyjnymi, a ich pierwszy zwarty opis został podany przez szwajcarskiego chemika Alfreda Wernera w roku 1893. Fakt, że orbitale d nie są zwrócone w określonym kierunku powoduje, że całki nakrywania są dużo mniejsze niż odpowiednie całki dla orbitali p. Wiele związków koordynacyjnych wykazuje dość wysoką symetrię, a te które jej nie przejawiają mogą być rozpatrywane w kategoriach wysokiej mikrosymetrii. Przykładowo związek kobaltu [Co(NH3)6]2+ zawiera cząsteczki amoniaku rozmieszczone w wierzchołkach oktaedru czyli posiada symetrię Oh. Podstawienie jednej cząsteczki amoniaku jonem chlorkowym, zachowuje oktaedryczny rozkład ligandów, ale obniży symetrię do C4v. Minmo to chociaż poziomy energetyczne zostają zaburzone to ich opis w oparciu o symetrię Oh jest nadal aktualny.
Geometria sfery koordynacyjnej
Jedną z konsekwencji przyjmowania przez związki koordynacyjne geometrii sfery koordynacyjnej jest zjawisko izomerii geometrycznej. Tego typu izomerii nie wykazują związki o liczbach koordynacyjnych 2 i 3 oraz tetraedryczne o liczbie koordynacyjnej 4. Związki o kwadratowym i oktaedrycznym wielościanie koordynacyjnym wykazują izomerię typu cis i trans.
 Izomeria cis i trans.W przypadku związków o geometrii oktaedru typu [MA3B3] występuje izomeria mer i fac
Izomeria cis i trans.W przypadku związków o geometrii oktaedru typu [MA3B3] występuje izomeria mer i fac
 Izomeria mer i fac związków o oktaedrycznym wielościanie koordynacyjnym.Kolejny rodzaj to izomeria optyczna, której przykłady dla związków o oktaedrycznym wielościanie koordynacyjnym zostały przedstawione na schemacie:
Izomeria mer i fac związków o oktaedrycznym wielościanie koordynacyjnym.Kolejny rodzaj to izomeria optyczna, której przykłady dla związków o oktaedrycznym wielościanie koordynacyjnym zostały przedstawione na schemacie:
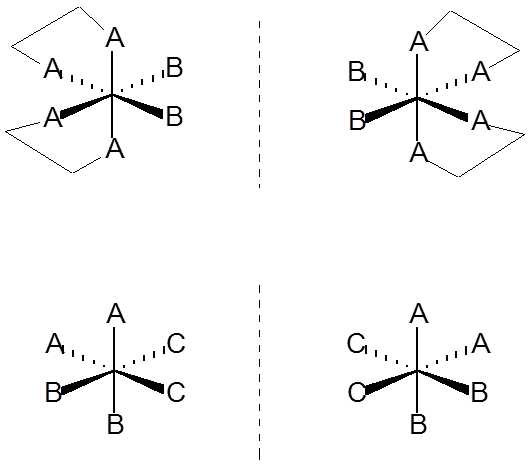 Kolejne rodzaje izomerii związków koordynacyjnych to izomeria konstytucyjna obejmująca:
Kolejne rodzaje izomerii związków koordynacyjnych to izomeria konstytucyjna obejmująca:
- izomerię hydratacyjną
[Cr(H2O)4Cl2]Cl•2H2O – zielony
[Cr(H2O)5Cl]Cl2•H2O – niebieskozielony
[Cr(H2O)6]Cl3 – fioletowy
- jonizacyjną
[Co(NH3)5Cl]SO4 – ciemnofioletowy
[Co(NH3)5SO4]Cl – fioletowoczerwony
- koordynacyjną
[Co(NH3)6][Cr(CN)6] i [Cr(NH3)6][Co(CN)6]
oraz izomerię wiązania:
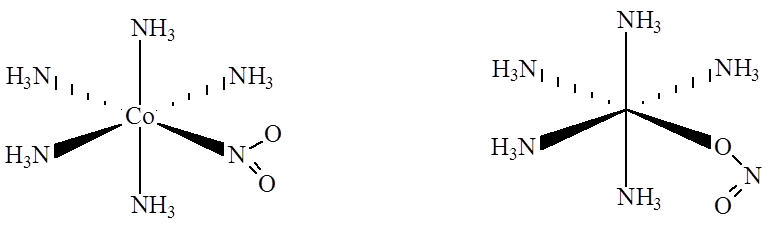 Struktura elektronowa związków koordynacyjnych
Struktura elektronowa związków koordynacyjnych
Diagramy orbitali cząsteczkowych dla związków koordynacyjnych o geometrii oktaedrycznej i symetrii Oh są analogiczne do zaprezentowanego wcześniej diagramu dla cząsteczki SF6. Dla związku, w którym ligandy nie tworzą wiązań π z metalem, orbitale t2g są niewiążące i głównie zlokalizowane na atomie centralnym. Podobnie antywiążący poziom eg powstaje z orbitali d atomu centralnego. W związku z tym w tym poziomie elektronowym w równym stopniu uczestniczą orbitale metalu i ligandów. Wiążące orbitale typu σ w skład których wchodzą orbitale eg, a1g i t1u, mają w dużym stopniu charakter orbitali ligandów. Schematycznie można to przedstawić następująco:
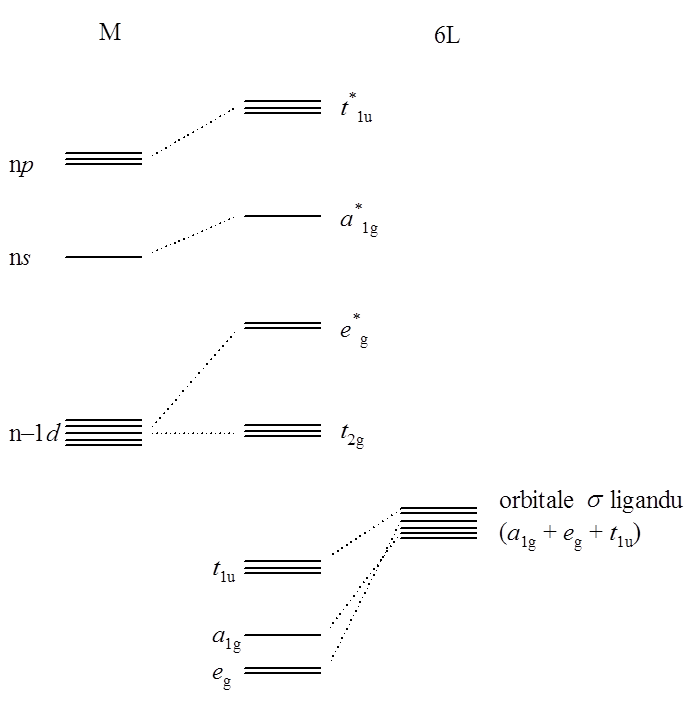 W przypadku gdy ligandy tworzą z atomem centralnym wiązania σ i π diagram orbitali cząsteczkowych ulega zmianie. Weźmy pod uwagę ligandy chlorkowe. Jeden z orbitali 3p chloru ma symetrię σ, a dwa pozostałe symetrię π względem osi metal–chlor. W związku z tym wiązanie pomiędzy metalem a chlorem zawierające te orbitale będzie wiązaniem typu π. W płaszczyźnie xz można to przedstawić schematycznie w sposób następujący:
W przypadku gdy ligandy tworzą z atomem centralnym wiązania σ i π diagram orbitali cząsteczkowych ulega zmianie. Weźmy pod uwagę ligandy chlorkowe. Jeden z orbitali 3p chloru ma symetrię σ, a dwa pozostałe symetrię π względem osi metal–chlor. W związku z tym wiązanie pomiędzy metalem a chlorem zawierające te orbitale będzie wiązaniem typu π. W płaszczyźnie xz można to przedstawić schematycznie w sposób następujący:
Podobnie tworzą się wi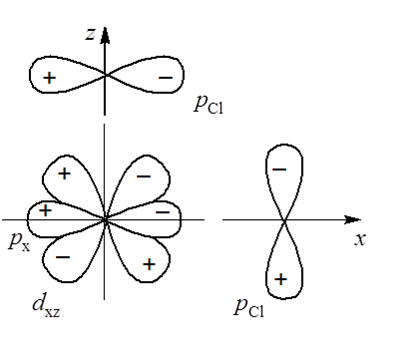 ązania typu π dla pozostałych osi. Orbitale typu π liganda przekształcają się jako t1u + t1g+ t2u + t2g. Orbitale p przekształcają się jako t1u, a orbitale d metalu jako eg + t2g. Ze względu na symetrię można oczekiwać oddziaływania orbitali d metalu z orbitalami π liganda. Natomiast charakter tego oddziaływania uzależniony jest od energii orbitali liganda. W przypadku chloru orbitale p są silnie związane, ale przykładowo cząsteczka CO posiada nisko energetyczne, antywiążące orbitale o symetrii π. W związku z tym struktura elektronowa związków koordynacyjnych z ligandami chlorowymi i karbonylowymi będzie różna. W związkach z ligandami posiadającymi orbitale π o niskiej energii, następuje zwiększenie energii orbitali cząsteczkowych t1u i t2g, i w tym przypadku mają one głównie charakter metalu i stają się antywiążącymi orbitalami cząsteczkowymi π. Orbitale t1g i t2u stają się niewiążące. Istotna zmiana dotyczy różnicy energii pomiędzy poziomami eg i t2g czyli parametru rozszczepienia Δ, oznaczanego także symbolem 10Dq. W rozpatrywanym przypadku następuje obniżenie jego wartości z powodu zwiększenia energii orbitali t2g metalu i przeniesienia części ładunku z zajętych orbitali π liganda na orbitale d metalu.
ązania typu π dla pozostałych osi. Orbitale typu π liganda przekształcają się jako t1u + t1g+ t2u + t2g. Orbitale p przekształcają się jako t1u, a orbitale d metalu jako eg + t2g. Ze względu na symetrię można oczekiwać oddziaływania orbitali d metalu z orbitalami π liganda. Natomiast charakter tego oddziaływania uzależniony jest od energii orbitali liganda. W przypadku chloru orbitale p są silnie związane, ale przykładowo cząsteczka CO posiada nisko energetyczne, antywiążące orbitale o symetrii π. W związku z tym struktura elektronowa związków koordynacyjnych z ligandami chlorowymi i karbonylowymi będzie różna. W związkach z ligandami posiadającymi orbitale π o niskiej energii, następuje zwiększenie energii orbitali cząsteczkowych t1u i t2g, i w tym przypadku mają one głównie charakter metalu i stają się antywiążącymi orbitalami cząsteczkowymi π. Orbitale t1g i t2u stają się niewiążące. Istotna zmiana dotyczy różnicy energii pomiędzy poziomami eg i t2g czyli parametru rozszczepienia Δ, oznaczanego także symbolem 10Dq. W rozpatrywanym przypadku następuje obniżenie jego wartości z powodu zwiększenia energii orbitali t2g metalu i przeniesienia części ładunku z zajętych orbitali π liganda na orbitale d metalu.
W drugim przypadku, ligandów posiadających puste orbitale π o stosunkowo niskiej energii, jak w przypadku cząsteczki CO, nastapi nakładanie orbitali t2g liganda z orbitalami t2g metalu. Ponieważ orbital t2g metalu ma niższą energię to powstający wiążący orbital cząsteczkowy będzie miał charakter orbitalu metalu z uwzględnieniem delokalizacji elektronów na ligandach. Ponieważ orbital t2g jest teraz wiążący, to wartość Δ jest większa niż w poprzednim przypadku. Jest to związane z akceptorowymi właściwościami liganda karbonylowego, który skoordynowany do metalu akceptuje ładunek an antywiążące orbitale π*, co przejawia się w wydłużeniu wiązania C–O i przesunięciu maksimum pasma częstości drgań grupy karbonylowej z 2189 cm–1 w przypadku tlenku węgla do około 2000 cm–1 obserwowanej dla karbonylowych związków koordynacyjnych. Schematycznie poziomy energetyczne orbitali cząsteczkowych dla obydwu przypadków można przedstawić następująco:
 i akceptorami π (prawy)") Diagram orbitali cząsteczkowych dla związku koordynacyjnego z ligandami będącymi donorami p (lewy) i akceptorami p (prawy)Wiązania typu π mogą wystąpić wtedy gdy ligand będący donorem π może domieszać pewien udział orbitali t2g metalu do swoich zajętych orbitali π, co powoduje obniżenie wartości Δ i prowadzi do przeniesienia gęstości elektronowej do metalu, lub też w przypadku gdy ligand posiadający wolne antywiążące orbitale π na które może akceptować ładunek z orbitali t2g metalu, co zwiększa wartość parametru Δ. Takie zachowanie ligandów i jego wpływ na strukturę elektronową związków koordynacyjnych powoduje, że związki z ligandami o właściwościach π–akceptorowych powinny być związkami o niskim spinie ze względu na dużą wartość parametru rozszczepienia. Natomiast związki koordynacyjne z ligandami będącymi silnymi donorami π, tak jak jony fluorkowe, luba ligandami będącymi słabymi donorami σ, będą przejawiały tendencję do tworzenia układów o wysokim spinie. Dalszą konsekwencją jest to, że związki, w których występują zajęte orbitale t2g będą trwalsze gdy ligandami są akceptory π (poziom t2g jest wtedy wiążący), a mniej trwałe gdy ligandami są silne σ–donory (poziom t2g jest niewiążący) lub donory π, gdyż wtedy poziom t2g staje się antuwiążący. Inaczej mówiąc ligandy π–akceptorowe stabilizują niskie stopnie utlenienia metali. Niewiążący lub słabo antywiążący charakter elektronów d atomu centralnego w związkach koordynacyjnych tłumaczy różnorodność stopni utlenienia metali na jakich tworzą one związki koordynacyjne.
Diagram orbitali cząsteczkowych dla związku koordynacyjnego z ligandami będącymi donorami p (lewy) i akceptorami p (prawy)Wiązania typu π mogą wystąpić wtedy gdy ligand będący donorem π może domieszać pewien udział orbitali t2g metalu do swoich zajętych orbitali π, co powoduje obniżenie wartości Δ i prowadzi do przeniesienia gęstości elektronowej do metalu, lub też w przypadku gdy ligand posiadający wolne antywiążące orbitale π na które może akceptować ładunek z orbitali t2g metalu, co zwiększa wartość parametru Δ. Takie zachowanie ligandów i jego wpływ na strukturę elektronową związków koordynacyjnych powoduje, że związki z ligandami o właściwościach π–akceptorowych powinny być związkami o niskim spinie ze względu na dużą wartość parametru rozszczepienia. Natomiast związki koordynacyjne z ligandami będącymi silnymi donorami π, tak jak jony fluorkowe, luba ligandami będącymi słabymi donorami σ, będą przejawiały tendencję do tworzenia układów o wysokim spinie. Dalszą konsekwencją jest to, że związki, w których występują zajęte orbitale t2g będą trwalsze gdy ligandami są akceptory π (poziom t2g jest wtedy wiążący), a mniej trwałe gdy ligandami są silne σ–donory (poziom t2g jest niewiążący) lub donory π, gdyż wtedy poziom t2g staje się antuwiążący. Inaczej mówiąc ligandy π–akceptorowe stabilizują niskie stopnie utlenienia metali. Niewiążący lub słabo antywiążący charakter elektronów d atomu centralnego w związkach koordynacyjnych tłumaczy różnorodność stopni utlenienia metali na jakich tworzą one związki koordynacyjne.
Wielkość parametru rozszczepienia 10Dq dla większości związków koordynacyjnych zawiera się w zakresie 7000 <Δ< 30000 cm-1 czyli w przybliżeniu od 1 do 4 eV. Doświadczalnie stwierdzono, że ligandy można uporządkować w szereg według wzrastających wartości parametru rozszczepienia. Takie uporządkowanie nosi nazwę szeregu spektrochemicznego gdyż ułożono go posługując się danymi eksperymentalnymi pozyskanymi z widm związków koordynacyjnych w zakresie UV-VIS.
| małe wartości 10Dq | duże wartości 10Dq | |
| I– < Br– < S2– < SCN– < Cl– < F– < OH– < OH2 < CH3CN < NH3 ≈ py < PR3 < CN– < CO < NO+ | ||
| π–donory | σ–donory | π–akceptory |
Dla związków o geometrii oktaedrycznej o konfiguracji d4, d5, d6 i d7 mamy dwa możliwe sposoby obsadzenia orbitali poziomu energetycznego t2g i eg, z których jeden wymaga zachowania reguły Hunda (stan wysokospinowy). Drugi polega na wypełnieniu w całości poziomu t2g i dopiero potem poziomu eg przez co osiąga się stan niskospinowy. Wybór jednej z możliwości zależy od parametru 10Dq. Jeżeli różnica energii pomiędzy zdegenerowanymi orbitalami d jest dostatecznie duża to jony centralne uzyskują niskospinowe konfiguracje elektronowe, a jeżeli zbyt niskie – wysokospinowe. Zależność pomiędzy parametrem rozszczepienia a ligandami i atomem centralnym została przez Jørgensena zdefiniowana wzorem: 10Dq≈f(liganda).g(atomu centralnego) gdzie czynniki f i g są wartościami eksperymentalnymi. Podobnie wartość parametru B Racah można przedstawić zależnością B–B0(1–h.k); B0 – oznacza wartość parametru dla wolnego jonu. Przykładowe wartości parametrów występujących w zależnościach Jørgensena przestawiają się następująco:
| jon | g | k | ligand | f | h |
| Co2+ | 9,3 | 0,15 | acac | 1,0 | |
| Co3+ | 20,8 | 0,35 | Br– | 0,75 | 2,0 |
| Cr2+ | 13,0 | CH3COO | 0,96 | ||
| Cr3+ | 17,0 | 0,17 | (4)Cl– | –0,38 | 2,0 |
| Fe2+ | 10,0 | CN– | 1,5 | 2,0 | |
| Fe3+ | 14,0 | 0,24 | NCS– | 1,03 | |
| Mo3+ | 19,0 | 0,15 | en | 1,27 | 1,4 |
| Ni2+ | 8,5 | 0,12 | F– | 0,88 | 0,8 |
| Re4+ | 35 | H2O | 1,00 | ||
| Rh3+ | 27 | NH3 | 1,2 | 1,2 | |
| V2+ | 12,4 | 0,08 | OH– | 0,94 | |
| V3+ | 20,0 | 0,29 | ox | 0,98 | 1,5 |
acac= acetyloacetionian; en = etylenodiamina; ox = jon szczawianowy
Należy pamiętać, że podane wartości mają charakter przybliżony, a wartość g wykazuje zależność od okresu, w którym znajduje się dany pierwiastek: g(3dn):g(4dn):g(5dn) = 1:1,45:1,75 oraz od stopnia utlenienia: g(+2):g(+3):g(+4) = 1:1,6:1,9. Konsekwentnie wielkość parametru rozszczepienia zmienia się wraz z przejściem od czwartego do piątego i szóstego okresu. Wartości 10Dg, częściowo na skutek większych rozmiarów i silniejszej polaryzowalności, wzrastają o około 30% w szeregu 3d < 4d < 5d.
Związki o tetraedrycznej geometrii sfery koordynacyjnej
Dla związków o niezaburzonej geometrii tetraedrycznej wielościanu koordynacyjnego diagram poziomów orbitalnych można wyznaczyć analogicznie jak dla okaedrycznych związków koordynacyjnych. Orbitale σ ligandów przekształcają się jako a1 i t2 (brak symboli g i u jest wynikiem braku środka symetrii w tetraedrze, w odróżnieniu od oktaedru) i mogą oddziaływać z orbitalami s (a1), d (t2) oraz w mniejszym stopniu p (t2) atomu centralnego. Orbitale e są niewiążące, a powłoka d rozszczepia się na dwa poziomy e i t2. Przy czym orbitale t2 są wyżej energetyczne niż e, czyli kolejność rozszczepionych poziomów podpowłoki d ulega odwróceniu w stosunku do związków oktaedrycznych. Dodatkowo wielkość parametru rozszczepienia Δ w przypadku teotraedru jest znacznie mniejsza niż obserwowana dla oktaedrycznego wielościanu koordynacyjnego. Przyjmuje się, w oparciu o wyniku uzyskane przy rozpatrywaniu modelu jonowego, że jeżeli długości wiązań pozostają stałe to Δtetr = 4/9 Δokt.
Diagram poziomów orbitalnych dla związku o geometrii tetraedrycznej wielościanu koordynacyjnego jest następujący:
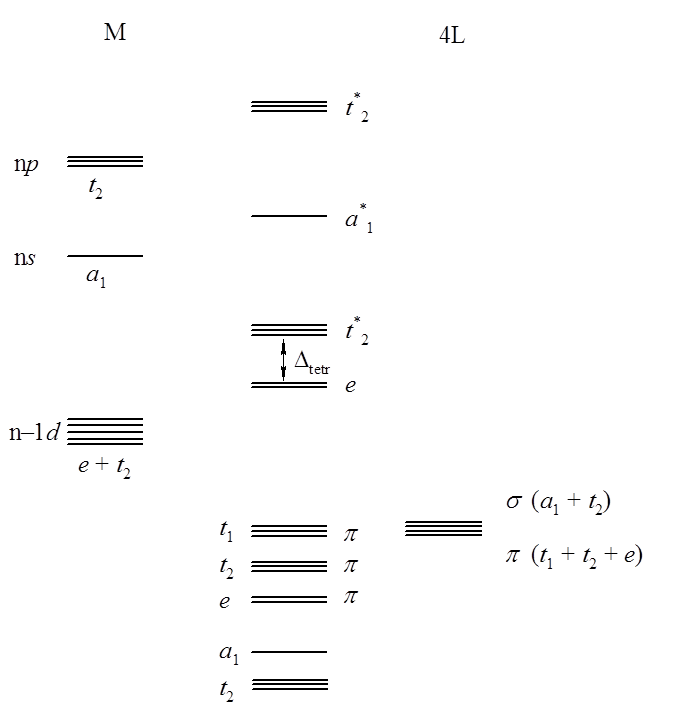 Ze względu na to, że wartość rozszczepienia Δtetr jest mniejsza niż analogiczna wartość dla oktaedru, a wielkość odpychania elektronowego pozostaje ta sama, wszystkie związki koordynacyjne o geometrii tetraedru są związkami wysokospinowymi. Nie ma wyróżnionej wartości obsadzenia elektronami orbitali d, dla której geometria tetraedryczna byłaby sprzyjają. Generalnie okatedryczne związki koordynacyjne wykazują większa stabilność od tetraedrycznych. Natomiast należy pamiętać o tym, że w przypadku niewielkich rozmiarów atomu centralnego oktaedryczny wielościan koordynacyjny może być niekorzystny ze względu na wzajemne odpychanie ligandów. ponadto w tetraedrze orbitale p i d atomu centralnego wykazują taką samą symetrię (t2) w związku z tym cząsteczkowy orbital t*2 zawiera domieszkę orbitali p metalu, co przejawia się przykładowo większą intensywnością przejść elektronowych e→t*2 w porównaniu do intensywności analogicznych przejść elektronowych dla związków o geometrii oktaedrycznej. Wynika to z faktu, że przejścia te mają w pewnym stopniu charakter przejść d→p, a takie nie są zabronione przez mechanizm dipola elektrycznego.
Ze względu na to, że wartość rozszczepienia Δtetr jest mniejsza niż analogiczna wartość dla oktaedru, a wielkość odpychania elektronowego pozostaje ta sama, wszystkie związki koordynacyjne o geometrii tetraedru są związkami wysokospinowymi. Nie ma wyróżnionej wartości obsadzenia elektronami orbitali d, dla której geometria tetraedryczna byłaby sprzyjają. Generalnie okatedryczne związki koordynacyjne wykazują większa stabilność od tetraedrycznych. Natomiast należy pamiętać o tym, że w przypadku niewielkich rozmiarów atomu centralnego oktaedryczny wielościan koordynacyjny może być niekorzystny ze względu na wzajemne odpychanie ligandów. ponadto w tetraedrze orbitale p i d atomu centralnego wykazują taką samą symetrię (t2) w związku z tym cząsteczkowy orbital t*2 zawiera domieszkę orbitali p metalu, co przejawia się przykładowo większą intensywnością przejść elektronowych e→t*2 w porównaniu do intensywności analogicznych przejść elektronowych dla związków o geometrii oktaedrycznej. Wynika to z faktu, że przejścia te mają w pewnym stopniu charakter przejść d→p, a takie nie są zabronione przez mechanizm dipola elektrycznego.
Związki o kwadratowej geometrii wielościanu koordynacyjnego
Związki koordynacyjne o geometrii płaskiego kwadratu przejawiają symetrię D4h. Rozpatrując wpływ wiązań π na rozkład orbitali cząsteczkowych związku o geometrii płaskiego kwadratu można stwierdzić, że ligandy π-akceptorowe obniżają energie niewiążących orbitali eg i b2g (wiązania σ), a ligandy π-donorowe będą energie tych orbitali zwiększać. Oczywiście wpływ ten silniej zaznacza się w przypadku orbitali dxy (b2g) niż dxz i dyz ze względu na większe nakładanie orbitali metalu i ligandów. Oddziaływanie p nie ma praktycznie wpływu na charakter orbitalu b1g. Uproszczony diagram dla omawianego przypadku przedstawiony został poniżej:
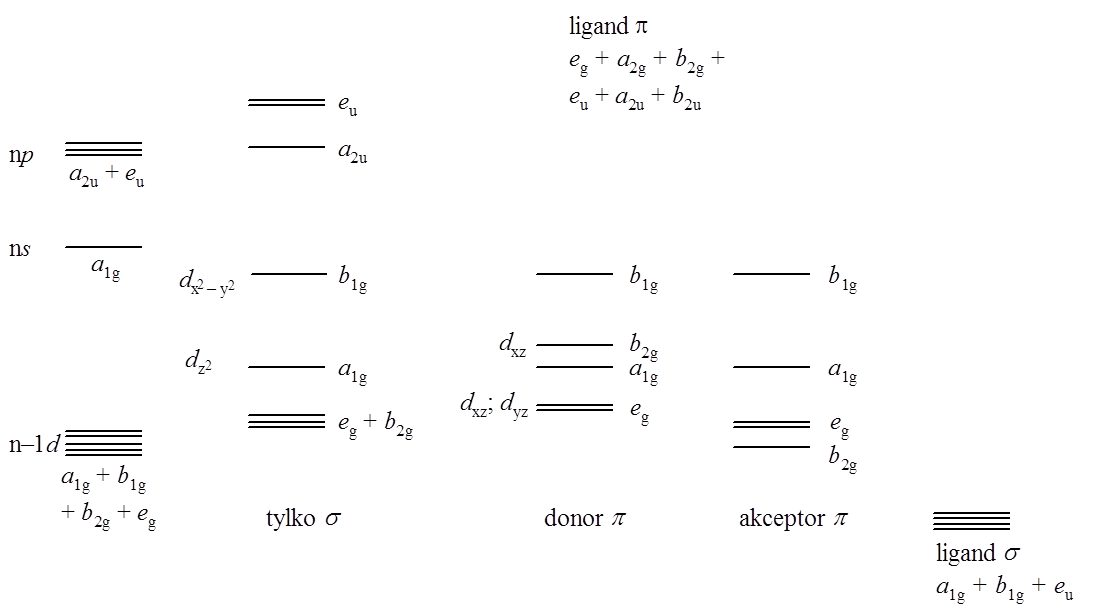 Jeżeli w cząsteczce związku o geometrii płaskiego kwadratu występują jedynie wiązania σ, to tylko orbitale dx2–y2 (b1g) i dz2 (a2g) mają odpowiednią symetrię aby nakładać się z orbitalami ligandów. Należy oczekiwać, że nakładanie orbitalu dx2–y2 będzie większe, ponieważ cały jest skierowany w stronę ligandów i dlatego powinien być silniej antywiążący. To tłumaczy fakt, że związki metali o konfiguracji d 8, posiadające zajęte cztery orbitale d wykazują tendencję do tworzenia struktur kwadratowych.
Jeżeli w cząsteczce związku o geometrii płaskiego kwadratu występują jedynie wiązania σ, to tylko orbitale dx2–y2 (b1g) i dz2 (a2g) mają odpowiednią symetrię aby nakładać się z orbitalami ligandów. Należy oczekiwać, że nakładanie orbitalu dx2–y2 będzie większe, ponieważ cały jest skierowany w stronę ligandów i dlatego powinien być silniej antywiążący. To tłumaczy fakt, że związki metali o konfiguracji d 8, posiadające zajęte cztery orbitale d wykazują tendencję do tworzenia struktur kwadratowych.
Biorąc pod uwagę moc donorowo-akceptorowa ligandów i rozpatrując deformację oktaedru polegająca na usunięciu ligandów w pozycjach aksjalnych połączoną ze skróceniem wiązań w płaszczyźnie ekwatorialnej (graniczny przypadek odkształcenia Jahna–Tellera) możemy przedstawić zmiany energii orbitali cząsteczkowych w postaci następującego diagramu:
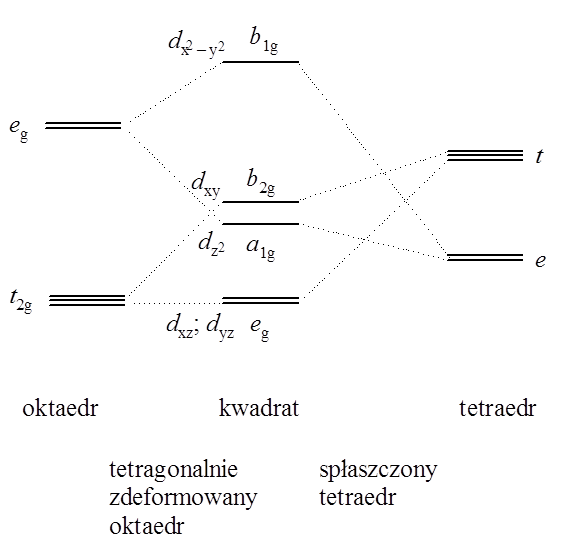 Bazując na tym diagramie można stwierdzić, że geometria kwadratowa związku koordynacyjnego dla metali o konfiguracji d 8 będzie korzystna jedynie wtedy gdy energia sparowania spinów będzie niższa niż rozszczepienie poziomów orbitali d w polu ligandów. Tylko w takim przypadku orbital dx2–y2 (b1g) pozostanie pusty. Innymi słowy kwadratowa geometria koordynacyjna jest charakterystyczna dla ligandów silnych pól, tworzących silnie kowalencyjne wiązana z atomem centralnym.
Bazując na tym diagramie można stwierdzić, że geometria kwadratowa związku koordynacyjnego dla metali o konfiguracji d 8 będzie korzystna jedynie wtedy gdy energia sparowania spinów będzie niższa niż rozszczepienie poziomów orbitali d w polu ligandów. Tylko w takim przypadku orbital dx2–y2 (b1g) pozostanie pusty. Innymi słowy kwadratowa geometria koordynacyjna jest charakterystyczna dla ligandów silnych pól, tworzących silnie kowalencyjne wiązana z atomem centralnym.
Dotychczasowe rozważania dotyczyły związków koordynacyjnych posiadających takie same ligandy. Jednak większość tego rodzaju związków charakteryzuje się tym, że w sferze koordynacyjnej metalu występuje więcej niż jeden rodzaj ligandu. Nie zmienia to jednak zasadności przedstawionych powyżej rozważań. Istotnym pojęciem staje się w tym wypadku zagadnienie mikrosymetrii. Jeżeli tylko ligandy w sferze koordynacyjnej nie różnią się zbytnio od siebie to układy typu MAxB6-x możemy traktować jako związki o geometrii oktaedrycznej. Jeżeli następuje rozszczepienie wewnątrz poziomów eg i t2g to jest ono mniejsze niż wartość różnicy energetycznej pomiędzy rozszczepionymi w oktaedrycznym polu ligandów poziomami eg i t2g. Oczywiście założenie, że lokalna symetria układu jest oktaedryczna jest dość zgrubnym przybliżeniem ale umożliwia łączenie poziomów eg z wiązaniami typu σ i poziomu t2g z wiązaniami π. Jest to bardzo użyteczne przybliżenie jeżeli rozważamy zagadnienia jakościowe, jednak należy pamiętać o tym przybliżeniu przy rozpatrywaniu konkretnych właściwości związków koordynacyjnych, zwłaszcza przy interpretacji ich widm elektronowych. Zwłaszcza nie należy wyciągać zbyt daleko idących wniosków w oparciu o przybliżenie lokalnej mikrosymetrii. szczególnie jest to istotne przy rozpatrywaniu właściwości magnetycznych związków koordynacyjnych, gdzie wymagane jest całościowe rozważenie struktury elektronowej związku.
Rozszczepienie poziomów orbitali d w polach o różnych symetriach przedstawia się następująco:
 Schemat rozszczepienia orbitali d w polach krystalicznych o różnych geometriachObniżenie symetrii pola krystalicznego powoduje zmianę degeneracji (orbitale zdegenerowane składają się z coraz mniejszej liczby orbitali atomowych) i co za tym idzie konieczność wprowadzenia innych poza 10Dq parametrów opisujących rozszczepienie orbitali d atomu centralnego. Najprostszym sposobem obniżenia symetrii oktaedrycznej jest zaburzenie tetragonalne lub trygonalne, czyli zmiana długości wiązań metal–ligand wzdłuż osi cztero– lub trójkrotnej (C4 lub C3). Zaburzenia te są reprezentowane poprzez wprowadzenie do równania Schrödingera odpowiednich operatorów opisujących oddziaływanie jonu centralnego z wytworzonym przez ligandy krystalicznym polem tetragonalnym lub trygonalnym. W konsekwencji poza parametrem rozszczepienia Dq pojawiają się dwa dodatkowe parametry oznaczane jako Ds i Dt w polu krystalicznym tetragonalnym i Dσ, Dτ w polu krystalicznym trygonalnym. Parametry te charakteryzują różnice miedzy ekwatorialnymi (podstawa wielościanu koordynacyjnego) i aksjalnymi polami krystalicznymi.
Schemat rozszczepienia orbitali d w polach krystalicznych o różnych geometriachObniżenie symetrii pola krystalicznego powoduje zmianę degeneracji (orbitale zdegenerowane składają się z coraz mniejszej liczby orbitali atomowych) i co za tym idzie konieczność wprowadzenia innych poza 10Dq parametrów opisujących rozszczepienie orbitali d atomu centralnego. Najprostszym sposobem obniżenia symetrii oktaedrycznej jest zaburzenie tetragonalne lub trygonalne, czyli zmiana długości wiązań metal–ligand wzdłuż osi cztero– lub trójkrotnej (C4 lub C3). Zaburzenia te są reprezentowane poprzez wprowadzenie do równania Schrödingera odpowiednich operatorów opisujących oddziaływanie jonu centralnego z wytworzonym przez ligandy krystalicznym polem tetragonalnym lub trygonalnym. W konsekwencji poza parametrem rozszczepienia Dq pojawiają się dwa dodatkowe parametry oznaczane jako Ds i Dt w polu krystalicznym tetragonalnym i Dσ, Dτ w polu krystalicznym trygonalnym. Parametry te charakteryzują różnice miedzy ekwatorialnymi (podstawa wielościanu koordynacyjnego) i aksjalnymi polami krystalicznymi.
Dalsze rozważania na temat właściwości cząsteczek związków koordynacyjnych znajdują się na stronach poświęconych teorii pola krystalicznego i teorii pola ligandów.