 Principles of Chemistry
Principles of Chemistry
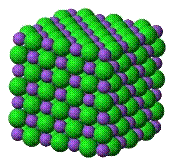 Wiązania jonowe
Wiązania jonowe
Model jonowy wiązania chemicznego stanowi najważniejsze z fenomenologicznych ujęć wiązania chemicznego. Substancję chemiczną traktuje się w tym modelu jako związek dwóch układów, z których jeden ma ładunek ujemny a drugi dodatni. Cząsteczkę w całości utrzymują oddziaływania elektrostatyczne wynikające z wzajemnego przyciągania dwóch różnoimiennych ładunków. Takie założenie pozwala przewidywać struktury związków, w których jony są otaczane przez jony o przeciwnym znaku tak ściśle jak jest to możliwe przy jednoczesnym odpychaniu jonów o tym samym znaku. W praktyce model jonowy znajduje zastosowanie jedynie w przypadku ciał stałych, chociaż może być stosowany w sposób jakościowy do roztworów i faz gazowych par jonowych.
Klasyczny przykład modelu jonowego stanowią halogenki litowców, w których wiązanie chemiczne powstaje w wyniku jonizacji atomu litowca z jednoczesnym przeniesieniem elektronu do atomu fluorowca. Taki proces generuje zysk energii elektrostatycznej sieci krystalicznej. Jednak nawet w przypadku atomu cezu mającego najsłabiej związany elektron walencyjny i atomu chloru, wykazującego najwyższe powinowactwo elektronowe, procesy uwalniania i przyłączania elektronu są endotermiczne. Mimo to zysk energetyczny wynikający z bardzo dużej energii elektrostatycznej uwolnionej podczas tworzenia kryształu jest większy niż energia potrzebna na jonizację i przyłączenie elektronu przez atomy. Cały proces można przedstawić w postaci cyklu termodynamicznego, zwanego cyklem Borna-Habera
 Cykl Borna-Habera dla procesu tworzenia soli jonowej MX. Wartości liczbowe w kJ/mol odnoszą się do CsCl.Energia uwolniona podczas zbliżania przeciwnie naładowanych jonów w trakcie tworzenia się kryształów to energia sieci i zgodnie z założeniami modelu jonowego jest to efekt stabilizacji sprzyjający tworzeniu wiązania chemicznego. Przyjmując, że jony wchodzące w skład cząsteczek ciała stałego są sztywnymi kulami, to znając strukturę ciała stałego możemy obliczyć energię sieci sumując wszystkie energie przyciągania i odpychania pomiędzy poszczególnymi jonami.. Dodatkowo przyjęcie modelu sztywnych kul umożliwia zastosowanie reguł stosunku promieni do przewidywania struktur i liczb koordynacyjnych. Ponieważ każdy jon znajduje się wewnątrz wielościanu koordynacyjnego, to najbardziej korzystne ułożenie nastąpi wówczas gdy atomy wielościanu koordynacyjnego będą we wzajemnym kontakcie. Jeżeli atom centralny będzie zbyt duży to koordynowane kule będą zmuszone do rozchodzenia się. Jeżeli będzie zbyt mały to korzystniejsze okaże się zmniejszenie liczby koordynacji, co pozwoli na ściślejsze wypełnienie przestrzeni wokół atomu (jonu) centralnego. Zastosowanie reguł geometrii w celu obliczenia stosunków promieni kationów i anionów, przy których jony "stykają się" prowadzi do następujących wyników:
Cykl Borna-Habera dla procesu tworzenia soli jonowej MX. Wartości liczbowe w kJ/mol odnoszą się do CsCl.Energia uwolniona podczas zbliżania przeciwnie naładowanych jonów w trakcie tworzenia się kryształów to energia sieci i zgodnie z założeniami modelu jonowego jest to efekt stabilizacji sprzyjający tworzeniu wiązania chemicznego. Przyjmując, że jony wchodzące w skład cząsteczek ciała stałego są sztywnymi kulami, to znając strukturę ciała stałego możemy obliczyć energię sieci sumując wszystkie energie przyciągania i odpychania pomiędzy poszczególnymi jonami.. Dodatkowo przyjęcie modelu sztywnych kul umożliwia zastosowanie reguł stosunku promieni do przewidywania struktur i liczb koordynacyjnych. Ponieważ każdy jon znajduje się wewnątrz wielościanu koordynacyjnego, to najbardziej korzystne ułożenie nastąpi wówczas gdy atomy wielościanu koordynacyjnego będą we wzajemnym kontakcie. Jeżeli atom centralny będzie zbyt duży to koordynowane kule będą zmuszone do rozchodzenia się. Jeżeli będzie zbyt mały to korzystniejsze okaże się zmniejszenie liczby koordynacji, co pozwoli na ściślejsze wypełnienie przestrzeni wokół atomu (jonu) centralnego. Zastosowanie reguł geometrii w celu obliczenia stosunków promieni kationów i anionów, przy których jony "stykają się" prowadzi do następujących wyników:
| rk/ra | > 0,732 | > 0,414 | > 0,225 |
| L.K. | 8 | 6 | 4 |
Pomimo tego, że związki o dużych wartościach stosunku rk/ra wykazują większe liczby koordynacyjne niż związki o mniejszych wartościach stosunku promieni kationu do anionu, to wiele związków dla których przewidywana jest wartość liczby koordynacyjnej 8 wykazu koordynację oktaedryczną (L.K=6). Jest to zrozumiałe jeżeli rozpatrzy się stronę elektrostatyczną problemu. Otóż zysk energii przy przejściu od struktury NaCl (L.K.=6) do struktury CsCl (L.K.=8) jest mniejszy niż 1%, a wtedy nieznaczny udział niejonowego efektu w wiązaniu może przeważyć w wyborze struktury kryształu i tym samym obalić przewidywania wynikające z reguły stosunków promieni. Staje się to zrozumiałe jeżeli rozpatrzymy charakter wiązania kowalencyjnego. Ten rodzaj oddziaływania zależy od stopnia wzajemnego nakładania orbitali, a tym samym korzystne jest takie ułożenie atomów, przy którym nastąpi jak największe nakładanie się orbitali atomów czy jonów. Stąd udział kowalencyjności w wiązaniu stabilizuje konkretną strukturę.
Moser i Pearson wykazali, na przykładzie szeregu związków typu AB pierwiastków zawierających w powłokach walencyjnych jedynie elektrony s i p i sumie elektronów walencyjnych równej 8 (NaCl, CaS, GaAs, Ge), że duże liczby koordynacyjne mają najczęściej atomy (jonu) w związkach ciężkich pierwiastków oraz w związkach o silnym wiązaniu jonowym. Dodatkowo wykazano, że w związkach o większym udziale wiązania jonowego występują ściśle upakowane szeregi kationów, a związki te mają zwarte struktury. Natomiast związki o większym udziale wiązania kowalencyjnego wykazują ścisłe upakowanie anionów i bardziej otwarte struktury. Zwiększenie liczby koordynacyjnej z 4 do 6 zwiększa trwałość sieci jonowej o 6–7%, a kowalencyjne oddziaływanie potrzebne do stabilizacji struktury o liczbie koordynacyjnej 4, w porównaniu ze strukturą o liczbie koordynacyjnej 6 będzie większe niż kowalencyjne oddziaływanie potrzebne do stabilizacji struktury o L.K.=6, w porównaniu ze strukturą o liczbie koordynacyjnej 8. Ze wzrostem stopnia kowalencyjności jest związany wzrost znaczenia nakładania się orbitali, w związku z tym wzrost stopnia kowalencyjności będzie faworyzował takie długości wiązań i takie liczby koordynacyjne, dla których nastąpi maksymalne nakładanie się orbitali. Wybór struktury zależy więc od dostępności orbitali, a to wiąże się również z ich energią. Krzem elementarny ma dostępne orbitale s i p i korzystniejsza będzie struktura tetraedryczna, odpowiadająca hybrydyzacji sp3. W izoelektronowej z krzemem cząsteczce NaCl kowalencyjność wiązania wynika z oddziaływania pomiędzy orbitalem 3s jonu sodu z obsadzonymi orbitalami 3p chloru. Największe nakładanie tych orbitali występuje w przypadku koordynacji oktaedrycznej. Orbital 3s chloru jest na tyle mocno związany (nisko energetyczny), że nie będzie stabilizowany przez jakiekolwiek oddziaływanie kowalencyjne. Udział kowalencyjności w wiązanie w NaCl będzie więc stabilizował koordynację oktaedryczną niż tetraedryczną. Tendencja do przejawiania dużych liczb koordynacyjnych występująca u cięższych pierwiastków odzwierciedla nie tylko większe rozmycie orbitali walencyjnych cięższych pierwiastków, co zmniejsza kierunkowość wiązań, lecz również zmniejszony udział orbitali s tych pierwiastków w wiązaniach Efekt ten nosi nazwę dehybrydyzacji.
Energia sieci
W modelu jonowym energię sieci przyjmuje się za czynnik decydujący o stabilizacji wiązania chemicznego. Podstawowe równanie na energię potencjalną sieci ma postać:
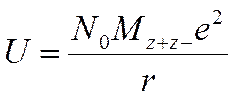 gdzie N0 –oznacza liczbę Avogadra, M – stałą Madelunga, z+ i z– – ładunki kationu i anionu, e –ładunek elektronu, r –najmniejszą odległość pomiędzy jonami o przeciwnym ładunku. Stała Madelunga odzwierciedla sumę oddziaływań typu przyciąganie i odpychanie pomiędzy jonami i zależy od od sposobu rozmieszczenia jonów i struktury kryształu. Wadami tego równania jest konieczność znajomości odległości pomiędzy jonami i struktury kryształu w celu oszacowania wartości stałej Madelunga. Nie znając dokładnej struktury kryształu można posłużyć się przybliżeniem wprowadzonym przez Kapustinskiego, zgodnie z którym przybliżona wartość stałej M można obliczyć mnożąc liczbę jonów w najmniejszej obojętnej jednostce związku przez 0,84.
gdzie N0 –oznacza liczbę Avogadra, M – stałą Madelunga, z+ i z– – ładunki kationu i anionu, e –ładunek elektronu, r –najmniejszą odległość pomiędzy jonami o przeciwnym ładunku. Stała Madelunga odzwierciedla sumę oddziaływań typu przyciąganie i odpychanie pomiędzy jonami i zależy od od sposobu rozmieszczenia jonów i struktury kryształu. Wadami tego równania jest konieczność znajomości odległości pomiędzy jonami i struktury kryształu w celu oszacowania wartości stałej Madelunga. Nie znając dokładnej struktury kryształu można posłużyć się przybliżeniem wprowadzonym przez Kapustinskiego, zgodnie z którym przybliżona wartość stałej M można obliczyć mnożąc liczbę jonów w najmniejszej obojętnej jednostce związku przez 0,84.
Równanie powyższe jest czystko elektrostatyczne i dodatkowo zakłada się w nim sztywność kul obrazujących jony. W związku z tym wprowadzono do niego poprawkę uwzględniającą odpychanie pomiędzy jonami o tych samych znakach, czyli odpychanie pomiędzy chmurami elektronowymi dwóch jonów, co przeciwdziała zetknięciu się jonów.
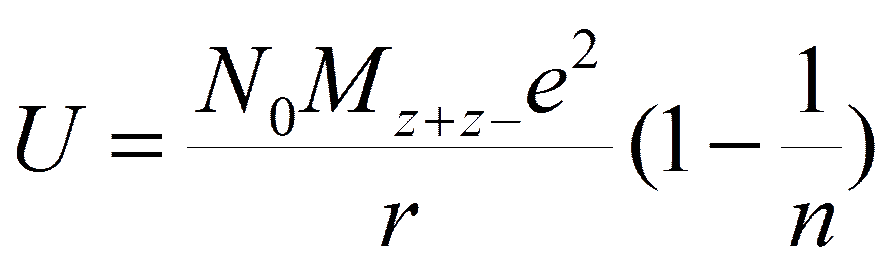
Wartość n wyznacza się z pomiarów ściśliwości ciał stałych, ale często stosuje się przybliżoną wartość równą 9.
Promienie jonowe wyznacza się kilkoma metodami. Obecnie najbardziej popularna jest metoda krystalograficzna. W przypadku dużych anionów i małych kationów przyjmuje się, że objętość komórki elementarnej zależy tylko od promieni anionów. Taką wartość jest stosunkowo łatwo obliczyć z pomiarów krystalograficznych i używa się jej jako stałą dla innych związków tego samego anionu. Pauling wykorzystał metodę półteoretyczną do wyznaczenia promieni jonowych polegającą na tym, że odległość pomiędzy dwoma przeciwnie naładowanymi jonami dzieli się zgodnie z ich efektywnymi liczbami atomowymi wyprowadzonymi na podstawie znajomości orbitali walencyjnych jonów. Zakłada się, że jony stykają się ze sobą. Kolejna metoda opiera się obliczeniach termochemicznych dotyczących energii sieci na podstawie danych eksperymentalnych.
Model jonowy potwierdza zgodność obliczanych wartości ciepeł tworzenia opartych na modelu Borna-Habera, ale również zgodność danych krystalograficznych z przewidywaniami opartymi na modelu jonowym. Potwierdzeniem modelu jonowego jest również fakt, że sole jonowe w ciele stałym są izolatorami elektrycznymi a przewodnictwo pojawia się dopiero po ich stopieniu. Wynika to z migracji jonów w stopionej soli. Niewielkie przewodnictwo soli stałych jest wynikiem drgań jonów w sieci krystalicznej lub obecności defektów w krysztale. Ponadto sole jonowe w większości są rozpuszczalne w rozpuszczalnikach polarnych, a w roztworach zachowują się jak jony przewodząc prąd elektryczny. Obserwowane efekty solwatacji zgadzają się z modelem jonowym.
Krytyka modelu jonowego
Przede wszystkim pomimo uproszczonego traktowania energii sieci i założeń przyjętych w tym modelu wyniki obliczonych ciepeł tworzenia znacznej grupy soli dobrze zgadzają się z wartościami eksperymentalnymi. Stanowi to na bardzo dobrą zgodność modelu z rzeczywistością. Podstawą modelu jonowego jest, że energia potrzebna do utworzenia jonów jest kompensowana przez energię sieci powstającego kryształu. W związku z tym związek KCl2 będzie nietrwały gdyż duża wartość drugiej energii jonizacji potasu nie jest równoważona przez zwiększoną energię sieci KCl2. Analogicznie sól CaCl byłaby nietrwała nie ze względu na skład, a na zachodzenie reakcji 2CaCl → Ca + CaCl2. W tym przypadku druga energia jonizacji jest kompensowana przez energię sieci, ale niestabilność CaCl wynikająca z możliwości przekształcenia się w inną sól jonową decyduje o powstawaniu odpowiedniej soli. Model jonowy może być stosowany nie tylko do jonów o zamkniętopowłokowej konfiguracji jonów, ale sprawdza się również dla jonów metali przejściowych. Natomiast, ponieważ w modelu tym stosuje się dane eksperymentalne nie jest on modelem teoretycznym, a ma charakter półempiryczny, co pozwala uniknąć problemów jakie w teorii orbitali cząsteczkowych napotykamy przy obliczaniu efektów odpychania elektronów i efektów korelacji. Należy sobie jednak zdawać sprawę z faktu, że nie istnieje czyste wiązanie jonowe, zawsze występuje pewien udział wiązania kowalencyjnego. Dobrym przykładem jest szereg halogenków srebra AgF, AgCl, AgBr, AgI. Dla tego szeregu obliczone w oparciu o model jonowy ciepła tworzenia są mniejsze niż wyznaczone eksperymentalnie, a eksperymentalne odległości między jonami wydają się zmniejszać w tym szeregu tak jakby jon srebra stawał się coraz mniejszy przy przechodzeniu od fluorku do jodku. Jest to związane z udziałem wiązania kowalencyjnego w tych solach. Fajans podał trzy reguły przewidywania kowalencyjnego charakteru połączeń chemicznych.
1. Gdy kation jest mały i ma duży ładunek, wówczas jest zdolny do ściślejszego przylegania do anionu i będzie przyciągał ładunek silniej niż anion. Jest to związane z charakterem kowalencyjnym wiązania. Przykładem są proste sole glinu zawierające, zgodnie z modelem jonowym Al3+ o promieniu 0,05 nm (sole te ulegają sublimacji). Dla odmiany sole lantanowców na +3 stopniu utlenienia o promieniu około 0,1 nm wykazują jonowy charakter w związkach.
2. Gdy anion jest duży i ma duży ładunek, wówczas im większy jest anion tym bardziej rozmyta jest chmura elektronowa, a w konsekwencji większe nakładanie orbitali anionu z orbitalami kationu. Z tego powodu cząsteczkę AgF dobrze opisuje model jonowy, który zawodzi przy opisie pozostałych halogenków srebra. Podobnie siarczki są bardziej kowalencyjne niż chlorki.
3. Gdy niektóre elektrony kationu są słabo ekranowane należy spodziewać się większej kowalencyjności wiązania. Przykładem jest porównanie soli magnezu(II) i cynku(II). Promienie jonowe obydwu jonów metali są zbliżone ale powinowactwo elektronowe Zn2+ jest większe niż w przypadku Mg2+, co wynika ze słabego ekranowania przez elektrony 3d Zn2+. Teoria orbitali cząsteczkowych pozwala oczekiwać większej delokalizacji ładunku w stronę jonów cynku(II), a tym samy większego udziału kowalencyjnego w tym przypadku niż w przypadku soli magnezu(II).
Model jonowy wyklucza całkowicie mieszanie się atomowych orbitali (hybrydyzacja) i zakłada, że nie występuje nakładanie orbitali dwóch jonów. Z ponktu widzenia teorii orbitali cząsteczkowych w trakcie oddziaływania dwóch jonów powstają dwa "orbitale cząsteczkowe" – obsadzony para elektronów orbital atomowy anionu fa i pusty orbital kationu fk. Energie tych orbitali różnią się od energii orbitali atomowych swobodnych atomów o energię elektrostatycznego oddziaływania jonów i energię odpychania elektronowego wynikającą z przemieszczenia elektronów. Pole kationu oddziałuje na orbital fa obniżając jego energię. Weźmy pod uwagę wyznacznik sekularny ![]() to wyrazy wyznacznika Hij poza przekątna są równe zero, a jedynie wyrazy na przekątnej (Hii) są przesuwane w wyniku oddziaływania dwóch pierwiastków. W takim wypadku spodziewamy się odchyleń od modelu jonowego zwłaszcza wtedy gdy wyrazy poza przekątna będą miały dużą wartość. Taka sytuacja ma miejsce gdy energie orbitali są zbliżone i należy spodziewać się ich dużego nakładania.
to wyrazy wyznacznika Hij poza przekątna są równe zero, a jedynie wyrazy na przekątnej (Hii) są przesuwane w wyniku oddziaływania dwóch pierwiastków. W takim wypadku spodziewamy się odchyleń od modelu jonowego zwłaszcza wtedy gdy wyrazy poza przekątna będą miały dużą wartość. Taka sytuacja ma miejsce gdy energie orbitali są zbliżone i należy spodziewać się ich dużego nakładania.
Energie solwatacji wykazują zbieżność z energiami sieci. Im mniejsze jony i im większy ich ładunek tym większa entalpia solwatacji. Jednak obliczanie rozpuszczalności soli jonowych na podstawie cyklu termodynamicznego należy mieć na uwadze fakt, że wyrazy odnoszące się do entalpii mogą mieć małą wartość i wtedy efekty związane z entropią zaczynają odgrywać decydującą rolę.