 Principles of Chemistry
Principles of Chemistry

Na początku był Arystoteles,
I obiekty w spoczynku pozostawały
w spoczynku
I obiekty w ruchu dążyły do spoczynku,
I wkrótce wszystko było w spoczynku,
I Bóg ujrzał, że to było nudne
I wtedy Bóg stworzył Bohra,
I powstała zasada,
A zasada była kwantowa
I wszystkie rzeczy były skwantowane,
Ale niektóre z nich wciąż były względne,
I Bóg ujrzał, że to było zagmatwane.
T. Joseph, Unified Field Theory
 Mechanika falowa
Mechanika falowa
Mechanika kwantowa postuluje, że każdy układ może być opisany za pomocą funkcji falowej, która jest funkcją wszystkich zmiennych tego układu. Wszystkie funkcje falowe interesujące z punktu widzenia chemii nieorganicznej muszą być rozwiązaniami równania Schrödingera:
![]() Rozważmy cząstkę poruszającą się swobodnie wzdłuż osi x. Ruch falowy tej cząstki można opisać funkcją falową typu: Ψ=Ψ(x,t). Najprostsza fala, to fala harmoniczna o takiej właściwości, że zwiększenie odległości x o λ, czyli długość fali, lub zwiększenie czasu t, o odwrotność częstości powoduje powtarzanie się takich samych właściwości Ψ. W tym przypadku funkcja Ψ zależy od x za pośrednictwem czynnika exp(±2πi kx), gdzie k oznacza liczbę falową czyli odwrotność długości fali k=1/λ. Zależność od czasu wyraża podobny czynnik równy exp(±2πivt), gdzie v jest częstością drgań. Ponieważ wyrażenie exp (i ω)=cosω + isinω, to obydwa człony wykładnicze są kombinacjami wyrazów sinusoidalnego i kosinusoidalnego. W ogólnym przypadku należy brać pod uwagę kombinacje wszystkich czterech możliwych iloczynów branych ze stałymi współczynnikami. Zwiększenie x o λ powoduje zwiększenie wykładnika o +2πi, czyli pomnożenie Ψ przez exp(±2πi ), czyli przez wartość 1. Równania jakie spełniają te wszystkie kombinacje możliwych iloczynów można wyznaczyć rozwiązując równania różniczkowe drugiego rzędu względem położenia x i czasu t:
Rozważmy cząstkę poruszającą się swobodnie wzdłuż osi x. Ruch falowy tej cząstki można opisać funkcją falową typu: Ψ=Ψ(x,t). Najprostsza fala, to fala harmoniczna o takiej właściwości, że zwiększenie odległości x o λ, czyli długość fali, lub zwiększenie czasu t, o odwrotność częstości powoduje powtarzanie się takich samych właściwości Ψ. W tym przypadku funkcja Ψ zależy od x za pośrednictwem czynnika exp(±2πi kx), gdzie k oznacza liczbę falową czyli odwrotność długości fali k=1/λ. Zależność od czasu wyraża podobny czynnik równy exp(±2πivt), gdzie v jest częstością drgań. Ponieważ wyrażenie exp (i ω)=cosω + isinω, to obydwa człony wykładnicze są kombinacjami wyrazów sinusoidalnego i kosinusoidalnego. W ogólnym przypadku należy brać pod uwagę kombinacje wszystkich czterech możliwych iloczynów branych ze stałymi współczynnikami. Zwiększenie x o λ powoduje zwiększenie wykładnika o +2πi, czyli pomnożenie Ψ przez exp(±2πi ), czyli przez wartość 1. Równania jakie spełniają te wszystkie kombinacje możliwych iloczynów można wyznaczyć rozwiązując równania różniczkowe drugiego rzędu względem położenia x i czasu t:
![]() i w tym wypadku podstawiając za iloczyn λ·v=u, czyli prędkość rozchodzenia się fali otrzymujemy:
i w tym wypadku podstawiając za iloczyn λ·v=u, czyli prędkość rozchodzenia się fali otrzymujemy:
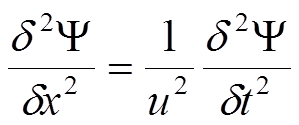 Równanie to jest klasycznym równaniem opisującym dowolny ruch falowy, w zależności od tego jakie znaczenie ma Ψ. Jednak równanie to nie może opisywać funkcji falowej Schrödingera, gdyż całka ∫ψ*ψdx byłaby zależna od czasu i unormowana funkcja falowa nie byłaby stale unormowana. Zależność od czasu nie występowałaby gdyby można było wybrać jeden z czynników, czyli albo exp(+2πivt ) albo exp(–2πivt ). Taka sytuacja miałaby miejsce gdyby równanie było pierwszego rządu, a nie drugiego mające dwa, a nie jedno rozwiązanie. Załóżmy jednak, że dopuszczalne jest tylko jedno rozwiązanie i dopuszczalny jest jedynie czynnik exp(–2πivt ). Wtedy znajdujemy rozwiązanie równania w postaci:
Równanie to jest klasycznym równaniem opisującym dowolny ruch falowy, w zależności od tego jakie znaczenie ma Ψ. Jednak równanie to nie może opisywać funkcji falowej Schrödingera, gdyż całka ∫ψ*ψdx byłaby zależna od czasu i unormowana funkcja falowa nie byłaby stale unormowana. Zależność od czasu nie występowałaby gdyby można było wybrać jeden z czynników, czyli albo exp(+2πivt ) albo exp(–2πivt ). Taka sytuacja miałaby miejsce gdyby równanie było pierwszego rządu, a nie drugiego mające dwa, a nie jedno rozwiązanie. Załóżmy jednak, że dopuszczalne jest tylko jedno rozwiązanie i dopuszczalny jest jedynie czynnik exp(–2πivt ). Wtedy znajdujemy rozwiązanie równania w postaci:
Ψ(x,t)= {A•exp(2πikx) + B•exp(–2πikx)}exp(–2πivt).
Podstawiając do równania zależności łączące pęd, długość fali i liczbę falową oraz częstość drgań z energią, czyli k = 1/λ = p/h, jak również ν =E/h, i wiedząc, że energia E = p2/2m + V, otrzymujemy:
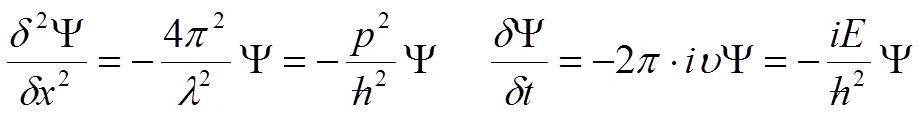 Z tych wzorów wynika następująca tożsamość:
Z tych wzorów wynika następująca tożsamość:
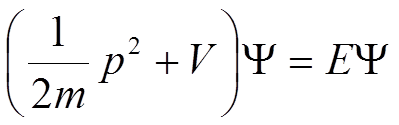 oraz równanie różniczkowe będące jednowymiarową postacią równania Schrödingera:
oraz równanie różniczkowe będące jednowymiarową postacią równania Schrödingera:
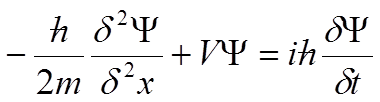 To równanie można również otrzymać rozpatrując klasyczne wyrażenie na energię jako funkcję położenia i pędu. Wyrażenie na energię zawiera operator Hamiltona, będący funkcją położenia i pędu:
To równanie można również otrzymać rozpatrując klasyczne wyrażenie na energię jako funkcję położenia i pędu. Wyrażenie na energię zawiera operator Hamiltona, będący funkcją położenia i pędu:
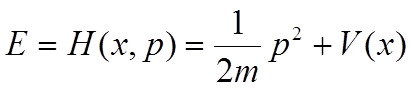 Teraz jeżeli w tym wyrażeniu pęd zastąpimy operatorem (ℏ/i )(δ/δx), a zamiast energii podstawimy operator (–ℏ/i )(δ/δt ) otrzymamy to samo równanie co powyżej. Rozpatrując cząstkę w trzech wymiarach musimy oczywiście zastosować operatory pędu wzdłuż trzech kierunków x, y , z.
Teraz jeżeli w tym wyrażeniu pęd zastąpimy operatorem (ℏ/i )(δ/δx), a zamiast energii podstawimy operator (–ℏ/i )(δ/δt ) otrzymamy to samo równanie co powyżej. Rozpatrując cząstkę w trzech wymiarach musimy oczywiście zastosować operatory pędu wzdłuż trzech kierunków x, y , z.
W opisie wiązań chemicznych dużą rolę odgrywają rozwiązania równania Schrödingera nazywane rozwiązaniami stacjonarnymi , czyli takimi, którym odpowiadają energia i gęstość ładunku niezależne od czasu. Takie stany opisują funkcje falowe o ogólnej postaci: ψ(x, y, z, t) = φ(x, y, z)·f(t). W takim wypadku otrzymujemy równanie:
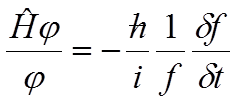 Obie strony tego równania są od siebie niezależne gdyż jedna jest funkcją położenia, a druga czasu. Obie będą sobie równe wtedy gdy każda z nich z osobna będzie równa pewnej, tej samej stałej, którą oznaczamy literą E. Jeżeli tak, to f(t) jest równe:
Obie strony tego równania są od siebie niezależne gdyż jedna jest funkcją położenia, a druga czasu. Obie będą sobie równe wtedy gdy każda z nich z osobna będzie równa pewnej, tej samej stałej, którą oznaczamy literą E. Jeżeli tak, to f(t) jest równe:
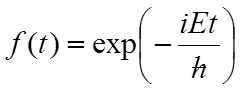 Natomiast niezależne od czasu równanie Schrödingera ma postać:
Natomiast niezależne od czasu równanie Schrödingera ma postać:
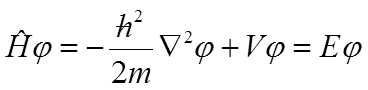 w którym ∇2φ oznacza:
w którym ∇2φ oznacza:
![]() Niezależne od czasu równanie Schrödingera rozwiązuje się ze względu na amplitudę φ(x, y, z) i następnie dołącza czynnik czasowy. Takie podejście oznacza, że różniczkowanie funkcji falowej względem czasu jest równoważne jej pomnożenie przez czynnik (–iE/ℏ), a w związku z tym E wyraża energię układu w stanie stacjonarnym. W takim ujęciu ewolucja czasowa jest opisywana zaburzeniem hamiltanianu, które określa się wprowadzając zmieniony hamiltonian i badając zachowanie funkcji falowej pod jego wpływem.
Niezależne od czasu równanie Schrödingera rozwiązuje się ze względu na amplitudę φ(x, y, z) i następnie dołącza czynnik czasowy. Takie podejście oznacza, że różniczkowanie funkcji falowej względem czasu jest równoważne jej pomnożenie przez czynnik (–iE/ℏ), a w związku z tym E wyraża energię układu w stanie stacjonarnym. W takim ujęciu ewolucja czasowa jest opisywana zaburzeniem hamiltanianu, które określa się wprowadzając zmieniony hamiltonian i badając zachowanie funkcji falowej pod jego wpływem.
Najprostszym układem jest atom wodoru. Ponieważ ładunek jądra jest równy e, a ładunek elektronu –-e. Elektron porusza się w przestrzeni trójwymiarowej i jego energia kinetyczna wynosi ½m(vx2 + vy2 + vz2). Energia potencjalna jest równa –e2/χ0r, gdzie r jest odległością elektronu od jądra. W takim wypadku równanie falowe ma postać:
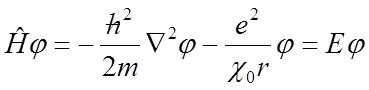 W przypadku atomu helu mamy dwa elektrony i równanie falowe uwzględnia wszystkie sześć zmiennych przestrzennych:
W przypadku atomu helu mamy dwa elektrony i równanie falowe uwzględnia wszystkie sześć zmiennych przestrzennych:
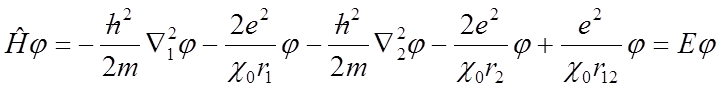 Po napisaniu równania falowego następnym zadaniem jest znalezienie takich wartości energii, dla których istnieją dopuszczalne funkcje falowe, czyli takie które są skończenie jednoznaczne i ciągłe, oraz mają ciągłe pierwsze pochodne i dają się unormować. Dla atomów wieloelektronowych stosuje się podejście Hartree’go, w którym ruch każdego elektronu rozważa się w uśrednionym polu pozostałych elektronów układu, czyli w uśrednionym polu jądra i wszystkich pozostałych elektronów. W polu Hartree’go energia potencjalna i-tego elektronu wynosi:
Po napisaniu równania falowego następnym zadaniem jest znalezienie takich wartości energii, dla których istnieją dopuszczalne funkcje falowe, czyli takie które są skończenie jednoznaczne i ciągłe, oraz mają ciągłe pierwsze pochodne i dają się unormować. Dla atomów wieloelektronowych stosuje się podejście Hartree’go, w którym ruch każdego elektronu rozważa się w uśrednionym polu pozostałych elektronów układu, czyli w uśrednionym polu jądra i wszystkich pozostałych elektronów. W polu Hartree’go energia potencjalna i-tego elektronu wynosi:
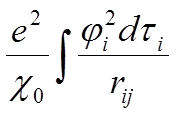 Po zsumowanie tych przyczynków i dodaniu członu –Ze2/χ0ri, pochodzącego od jądra, uśrednia się całość względem wszystkich kierunków uzyskując kulistosymetryczną funkcję energii potencjalnej. W praktyce rozwiązuje się te równania numerycznie, uzyskując w wyniku pierwsze przybliżenie funkcji falowej. Takie postępowanie prowadzi się w przypadku każdego elektronu uzyskując pole samouzgodnione z odpowiadającym mu rozkładem elektronów.
Po zsumowanie tych przyczynków i dodaniu członu –Ze2/χ0ri, pochodzącego od jądra, uśrednia się całość względem wszystkich kierunków uzyskując kulistosymetryczną funkcję energii potencjalnej. W praktyce rozwiązuje się te równania numerycznie, uzyskując w wyniku pierwsze przybliżenie funkcji falowej. Takie postępowanie prowadzi się w przypadku każdego elektronu uzyskując pole samouzgodnione z odpowiadającym mu rozkładem elektronów.
Ścisłe rozwiązanie równania falowego jest niemożliwe dla atomów zawierających więcej niż jeden elektron i oczywiście nie jest rozwiązywalne dla cząsteczek. Dla przykładu w cząsteczce metanu CH4 znajduje się pięć jąder i 10 elektronów, a tym samym pełne równanie falowe dla tej cząsteczki zawiera 45 zmiennych (3·15=45) Ścisłe rozwiązanie równania różniczkowego cząstkowego o tak wielu zmiennych jest niewykonalne. Problem upraszcza się stosując przybliżenia zakładające nieruchomość jąder. To przybliżenie jest uzasadnione gdyż masa jąder atomowych jest znacznie większa od masy elektronu (masa protonu jest większa od masy elektronu 1836 razy). Przybliżenie to zostało po raz pierwszy wprowadzone przez Borna i Oppenheimera w roku 1927 i jest niezwykle dokładne, a co istotniejsze jego przyjęcie czyli rozdzielenie ruchów jąder i elektronów uzasadnia pojęcia krzywej i powierzchni energii potencjalnej cząsteczki. Jednak przyjęcie przybliżenia Borna-Oppenheimera ogranicza liczbę zmiennych w stopniu nie wystarczającym do przeprowadzenia dokładnych obliczeń funkcji falowych cząsteczek. Dla metanu liczba zmiennych po przyjęciu tego przybliżenia zmniejsza się do 30, a to jeszcze zbyt wiele aby można było przeprowadzić dokładne obliczenia.
Metoda wariacyjna
Weźmy pod uwagę równanie Schrödingera i wyznaczmy z niego energię:
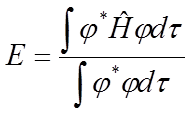 W przypadku funkcji unormowanej mianownik tego wyrażenia będzie równy jedności. Jeżeli znamy dokładną postać funkcji falowej φ to korzystając z tego równania obliczymy dokładną wartość energii. Jednak nie dysponujemy taką funkcją to zgodnie z tym co wykazał Rayleigh stosując przybliżoną funkcję falową uzyskujemy przybliżoną wartość energii układu, która będzie większa niż wartość dokładna. Metoda wariacyjna polega na dobieraniu odpowiednich parametrów funkcji falowej i na tej podstawie poszukiwaniu minimum energii.
W przypadku funkcji unormowanej mianownik tego wyrażenia będzie równy jedności. Jeżeli znamy dokładną postać funkcji falowej φ to korzystając z tego równania obliczymy dokładną wartość energii. Jednak nie dysponujemy taką funkcją to zgodnie z tym co wykazał Rayleigh stosując przybliżoną funkcję falową uzyskujemy przybliżoną wartość energii układu, która będzie większa niż wartość dokładna. Metoda wariacyjna polega na dobieraniu odpowiednich parametrów funkcji falowej i na tej podstawie poszukiwaniu minimum energii.
Metoda kombinacji linowych
Metoda kombinacji liniowych jest odmianą metody wariacyjnej szczególnie ważną z punktu widzenia chemii. W metodzie tej w ogólności możemy założyć, że dokładna funkcja falowa ψ ma cechy pewnych dwóch funkcji φ1 i φ2. W ogólności funkcje te nie muszą być rozwiązaniami żadnego równania falowego. Możemy teraz przedstawić funkcję ψ w postaci sumy przyczynków obydwu funkcji φ1 i φ2:
ψ = c1φ1 + c2φ2
Podejście takie jest uzasadnione na przykład gdy na elektron znajdujący się na orbitalu s działa pole elektromagnetyczne skierowane wzdłuż osi x. Wtedy pełna funkcja falowa takiego stanu nosi znamiona funkcji opisującej orbital s i jednocześnie funkcji opisującej orbital skierowany wzdłuż osi x czyli kierunku pola co wskazuje na udział orbitalu typu px..
Stałe w podanej powyżej sumie należy tak dobrać aby zminimalizować wyrażenie na energię. Wyrażenie na energię przybiera w tym ujęciu postać:
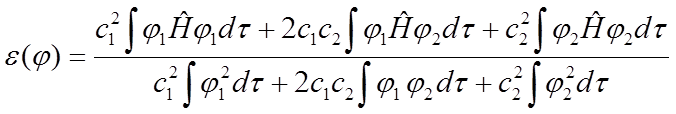 Symbolem ε(φ) oznaczono przybliżoną energię układu. Oznaczmy teraz odpowiednie całki symbolami Hij=∫φiĤφjdτ oraz Sij=∫φiφjdτ, przy czym ponieważ operator Ĥ jest hermitowski to Hij = Hji i Sij = Sji. Wielkość Sij nazywamy całką nakrywania i jest ona równa jedności gdy funkcje φij będą unormowane. Wielkości Hij są elementami diagonalnymi macierzy operatora Ĥ. Wyrażenie na energię będzie teraz wyglądało następująco:
Symbolem ε(φ) oznaczono przybliżoną energię układu. Oznaczmy teraz odpowiednie całki symbolami Hij=∫φiĤφjdτ oraz Sij=∫φiφjdτ, przy czym ponieważ operator Ĥ jest hermitowski to Hij = Hji i Sij = Sji. Wielkość Sij nazywamy całką nakrywania i jest ona równa jedności gdy funkcje φij będą unormowane. Wielkości Hij są elementami diagonalnymi macierzy operatora Ĥ. Wyrażenie na energię będzie teraz wyglądało następująco:
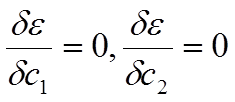 co prowadzi do równania wiekowego postaci:
co prowadzi do równania wiekowego postaci:
c1(H11 – ES11) + c2(H12 – ES12)=0
c1(H12 – ES12) + c2(H22 – ES22)=0
Równania te można przedstawić w postaci wyznacznika:
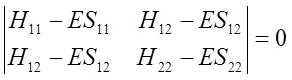 Wyznacznik jest utworzony ze współczynników znajdujących się przy niewiadomych c1 i c2 w równaniu na ψ i opisuje równanie kwadratowe, którego rozwiązaniem są dwie wartości energii E. Inaczej mówiąc po rozwiązaniu równania otrzymujemy energię stanu podstawowego i pierwszego stanu wzbudzonego układu opisanego funkcją falową ψ będącą kombinacją liniową dwóch funkcji φ.
Wyznacznik jest utworzony ze współczynników znajdujących się przy niewiadomych c1 i c2 w równaniu na ψ i opisuje równanie kwadratowe, którego rozwiązaniem są dwie wartości energii E. Inaczej mówiąc po rozwiązaniu równania otrzymujemy energię stanu podstawowego i pierwszego stanu wzbudzonego układu opisanego funkcją falową ψ będącą kombinacją liniową dwóch funkcji φ.
Interpretacja fizyczna φ
Pozostaje kwestia nadania sensu fizycznego funkcji falowej φ, która w mechanice falowej reprezentuje poruszającą się cząstkę. Przyjęto, że sama funkcja nie ma sensu fizycznego. natomiast kwadrat jej modułu, czyli wyrażenie |φ|2dτ jest równe prawdopodobieństwu znalezienia rozpatrywanej cząstki w elemencie objętości dτ. Można nieco inaczej zinterpretować funkcję falową. Weźmy pod uwagę elektron, który jest rozpostarty (zdelokalizowany) w postaci chmury ładunku, której gęstość w każdym punkcie jest wprost proporcjonalna do wartości |φ|2. W takim wypadku w miejscach gdzie wartość tego wyrażenia będzie największa chmura ładunku jest najgęstsza i w tym miejscu znajduje się najwięcej ładunku ujemnego. Różnica jaka występuje pomiędzy tymi dwoma podejściami jest subtelna ale ma duże znaczenie. Zamiast mówić o gęstości prawdopodobieństwa oznaczającej szansę znalezienia elektronu w danym elemencie przestrzeni dτ mamy rozmyty elektron o pewnej gęstości czyli ilości elektronów (ładunku) na jednostkę objętości. Z równania:
∫|φ(x,y,z)|2dτ = 1,
wynika, że całkowity ładunek elektronu, określony całką z gęstości po całej przestrzeni, jest równy dokładnie ładunkowi jednego elektronu. Pozostając przy poprzedniej interpretacji napotykamy trudność polegającą na tym, że elektron jako cząstka nie może rozpościerać się w przestrzeni o rozmiarach atomu czy cząsteczki. To pociąga za sobą jeszcze jeden istotny element. Otóż interpretacja probabilistyczna (oparta na prawdopodobieństwie) jest jedyną, która jest słuszna. Gdybyśmy byli w stanie dokładnie oznaczać położenie elektronu w atomie, to po wykonaniu szeregu takich pomiarów uzyskalibyśmy obraz chmury, w której najgęściej ułożone punkty pomiarowe odpowiadałyby miejscom gdzie prawdopodobieństwo znalezienia elektronu było najwyższe. Innymi słowy gęstość chmury ładunku stanowi miarę funkcji prawdopodobieństwa.
Na kolejnych stronach tego działu przedstawione powyżej zagadnienia zostały omówione w sposób bardziej szczegółowy, pozwalający na ich pełniejsze zrozumienie.