 Principles of Chemistry
Principles of Chemistry
 Oddziaływania międzycząsteczkowe
Oddziaływania międzycząsteczkowe
Znaczenie oddziaływań międzycząsteczkowych jest niezwykle istotne przy rozpatrywaniu właściwości materii, a ich rola jest równie ważna co wiązań chemicznych. W przypadku gazów to właśnie oddziaływania międzycząsteczkowe są powodem ich odstępstw od "doskonałości. Struktura cieczy, ich właściwości fizyczne takie jak ciepło parowania, napięcie powierzchniowe czy lepkość są uwarunkowane energią oddziaływań międzycząsteczkowych. W ciałach stałych odpowiadają one za szereg właściwości związanych również ze strukturą. Jeden rodzaj oddziaływań międzycząsteczkowych – wiązanie wodorowe – został już omówiony w części poświęconej wiązaniu chemicznemu. W tym miejscu ograniczymy się do oddziaływań o charakterze elektrostatycznym. Najsilniejsze tego typu oddziaływania występują pomiędzy dwoma różnoimiennymi jonami cząsteczkowymi. Od wiązań jonowych różni je to, że ładunek jest tutaj rozmieszczony na całej cząsteczce, a nie zlokalizowany na danym atomie. Siła oddziaływania pomiędzy jonami jest proporcjonalna do 1/r2. Siła tego oddziaływania zależy od ośrodka, w którym znajdują się jony gdyż efekty solwatacyjne wpływają na jego moc.
Oddziaływanie pomiędzy cząsteczkami posiadającymi trwałe momenty dipolowe ma również charakter elektrostatyczny, ale jego siła jest mniejsza niż w przypadku jonów gdyż mamy tutaj do czynienia z ładunkami cząstkowymi. Cząsteczka posiadająca trwały moment dipolowy może indukować moment dipolowy w cząsteczce elektrycznie obojętnej elektrycznie, i w tym wypadku pojawia się pomiędzy nimi oddziaływanie van der Waalsa. Siła tego oddziaływania jest proporcjonalna do 1/r6. Najsłabsze oddziaływania o charakterze elektrostatycznym pomiędzy cząsteczkami, występujące pomiędzy dipolami indukowanymi noszą nazwę oddziaływań dyspersyjnych lub też oddziaływań Londona. Oddziaływania te wynikają z faktu, że rozkład elektronów wokół jadra atomowego w danej chwili nie jest symetryczny, co powoduje pojawienie się chwilowego momentu dipolowego, który może indukować na sąsiednich atomach chwilowe indukowane momenty dipolowe. Oddziaływania dyspersyjne są bardzo słabymi oddziaływaniami ale maja istotne znaczenie dla cząsteczek związków niepolarnych i nie posiadających trwałych momentów dipolowych. Na ich moc ma wpływ ilość elektronów w cząsteczce i jej kształt. Większa chmura elektronowa, jak i większy dodatni ładunek jądra pozwalają uzyskać większy chwilowy moment dipolowy. Chwilowe momenty dipolowe w cząsteczkach o kształcie liniowym mogą znajdować się bliżej siebie, przez co oddziaływania te są silniejsze. W cząsteczkach sferycznych oddziaływania momentów dipolowych są słabsze niż w liniowych ze względu na większe odległości.
Oddziaływania warstwowe
Oddziaływania warstwowe pomiędzy elektronami π układów aromatycznych należą, łącznie z wiązaniami wodorowymi, do szeroko rozpowszechnionych oddziaływań występujących tak w układach biologicznych jak i w syntezowanych cząsteczkach związków chemicznych. Ten rodzaj oddziaływań ma znaczący wpływ na strukturę krystaliczną związków aromatycznych, w pewnym stopniu odpowiada za stereoselektywność reakcji w syntezie organicznej i odgrywa rolę w układach biologicznych na poziomie kwasów nukleinowych. Koncepcja oddziaływania pierścieni aromatycznych została po raz pierwszy przedstawiona dla wyjaśnienia budowy dimerów benzenu, w których dwie cząsteczki tego związku, w fazie gazowej, znajdują się w odległości 4.96 Å, a energia wiązania została oszacowana na 2 – 3 kcal/mol. Ze względu na bardzo słabe oddziaływanie trudno jest je badać w fazie gazowej, natomiast analiza krystalograficzna pozwoliła na dokładne określenie geometrii tego rodzaju oddziaływania, jak to zostało przedstawione na schemacie.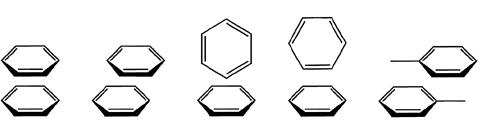 Dwie najkorzystniejsze konfiguracje układów aromatycznych to takie gdzie pierścienie aromatyczne są ustawione równolegle lub prostopadle w stosunku do siebie. Przy czym korzystniejsza energetycznie jest konfiguracja w kształcie litery T pierścieni aromatycznych. Ten rodzaj oddziaływania nie ma charakteru czysto elektrostatycznego, chociaż jeden z pierwszych modeli postulował oddziaływanie pomiędzy układem sprzężonym elektronów π pierścienia i elektronami σ wiązań C–H. Ponieważ cząsteczka benzenu nie posiada momentu dipolowego, wykazuje natomiast znaczny moment kwadrupolowy, należy przy rozpatrywaniu tego oddziaływania uwzględnić wzajemne przyciąganie i odpychanie momentów kwadrupolowych i wpływ sił Londona. Wiązania C–H wykazują lokalne momenty dipolowe, które powodują, że na nich zlokalizowany jest cząstkowy ładunek dodatni i odpowiedni ładunek ujemny ponad i pod płaszczyzną pierścienia. W związku z tym równoległe ułożenie pierścieni aromatycznych jest stabilizowane siłami dyspersyjnymi Londona i destabilizowane przez wzajemne oddziaływanie kwadrupolowe. To tłumaczy większą stabilność prostopadłej konfiguracji dimeru w stosunku do równoległego ułożenia cząsteczek benzenu. Takie przedstawienie sił działających w układach warstwowych wyjaśnia również wzajemne przesunięcie pierścieni przy równoległym ułożeniu cząsteczek oddziałujących związków. Łatwiej to zrozumieć gdy przedstawimy rozkład ładunku (momentu kwadrupolowego) na cząsteczkach związków uwzględniając wpływ podstawników stabilizujących lub destabilizujących układ aromatyczny.
Dwie najkorzystniejsze konfiguracje układów aromatycznych to takie gdzie pierścienie aromatyczne są ustawione równolegle lub prostopadle w stosunku do siebie. Przy czym korzystniejsza energetycznie jest konfiguracja w kształcie litery T pierścieni aromatycznych. Ten rodzaj oddziaływania nie ma charakteru czysto elektrostatycznego, chociaż jeden z pierwszych modeli postulował oddziaływanie pomiędzy układem sprzężonym elektronów π pierścienia i elektronami σ wiązań C–H. Ponieważ cząsteczka benzenu nie posiada momentu dipolowego, wykazuje natomiast znaczny moment kwadrupolowy, należy przy rozpatrywaniu tego oddziaływania uwzględnić wzajemne przyciąganie i odpychanie momentów kwadrupolowych i wpływ sił Londona. Wiązania C–H wykazują lokalne momenty dipolowe, które powodują, że na nich zlokalizowany jest cząstkowy ładunek dodatni i odpowiedni ładunek ujemny ponad i pod płaszczyzną pierścienia. W związku z tym równoległe ułożenie pierścieni aromatycznych jest stabilizowane siłami dyspersyjnymi Londona i destabilizowane przez wzajemne oddziaływanie kwadrupolowe. To tłumaczy większą stabilność prostopadłej konfiguracji dimeru w stosunku do równoległego ułożenia cząsteczek benzenu. Takie przedstawienie sił działających w układach warstwowych wyjaśnia również wzajemne przesunięcie pierścieni przy równoległym ułożeniu cząsteczek oddziałujących związków. Łatwiej to zrozumieć gdy przedstawimy rozkład ładunku (momentu kwadrupolowego) na cząsteczkach związków uwzględniając wpływ podstawników stabilizujących lub destabilizujących układ aromatyczny.
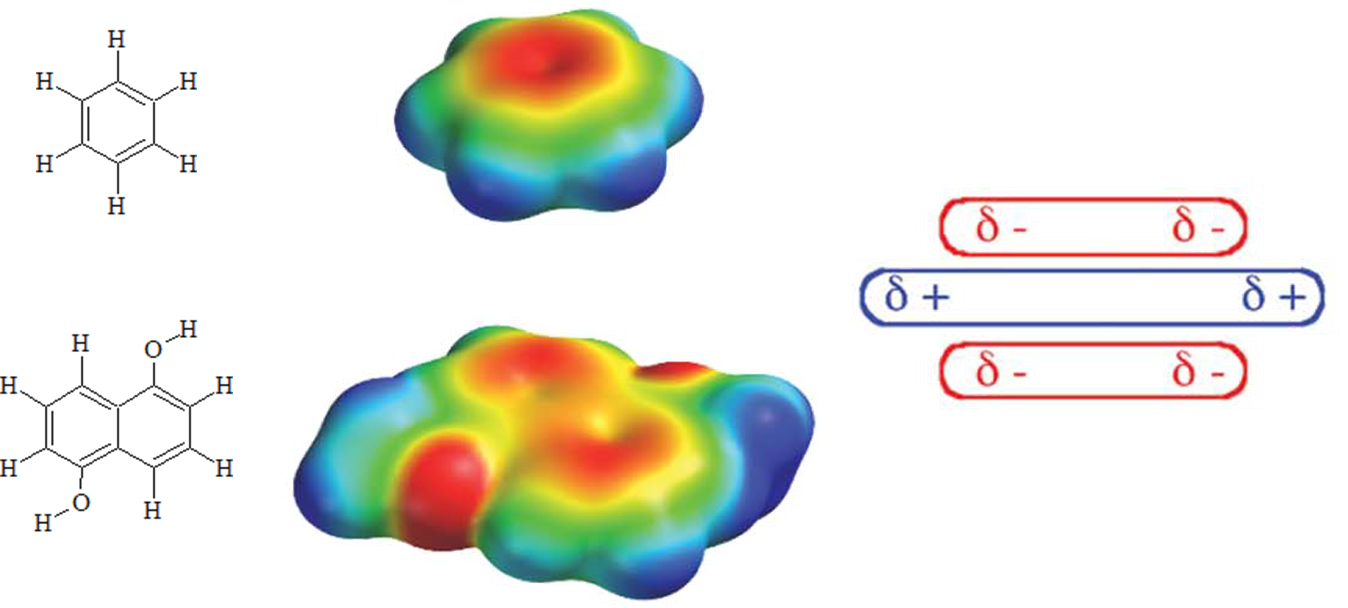
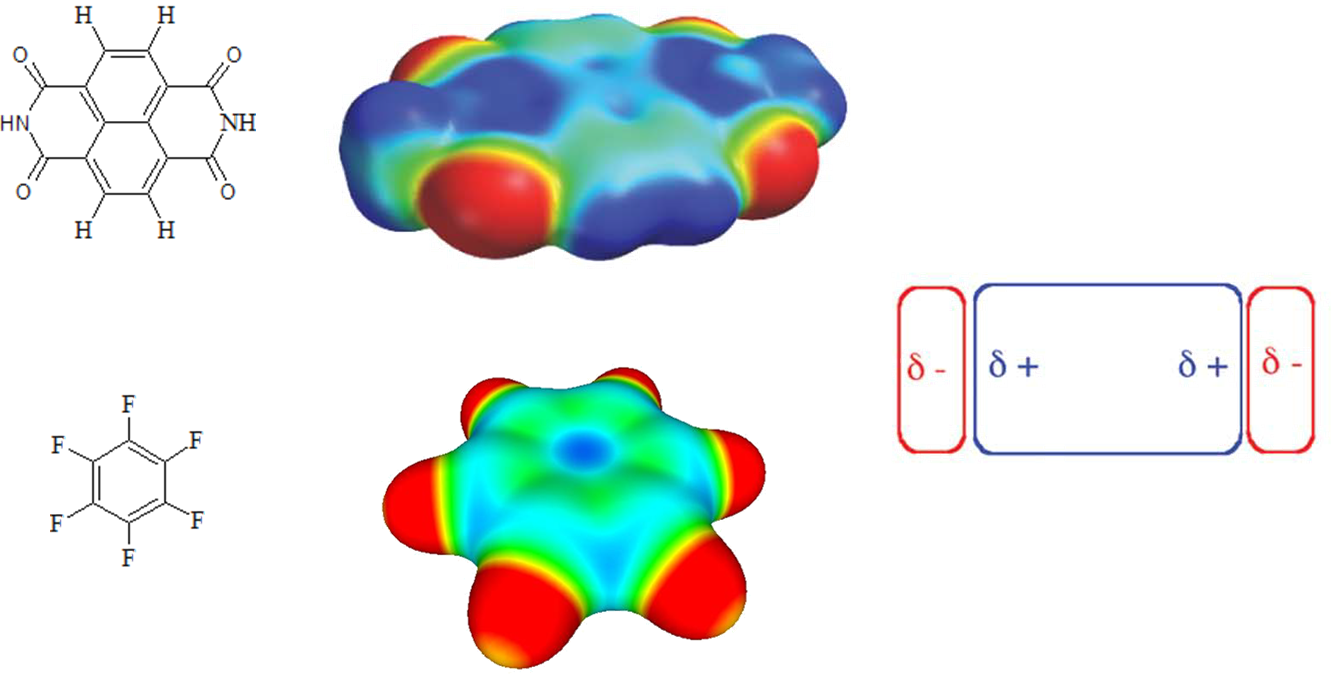 Jak łatwo zauważyć silnie destabilizujące układ aromatyczny podstawniki powodują zmianę rozkładu ładunku na cząsteczce związku, co ma wpływ na układ (geometrię) oddziaływujących cząsteczek. Weźmy pod uwagę dwie cząsteczki benzenu lub 1,5-dihydroksynaftalenu, w przypadku których mamy duży ładunek cząstkowy zlokalizowany nad i pod płaszczyzną pierścienia.
Jak łatwo zauważyć silnie destabilizujące układ aromatyczny podstawniki powodują zmianę rozkładu ładunku na cząsteczce związku, co ma wpływ na układ (geometrię) oddziaływujących cząsteczek. Weźmy pod uwagę dwie cząsteczki benzenu lub 1,5-dihydroksynaftalenu, w przypadku których mamy duży ładunek cząstkowy zlokalizowany nad i pod płaszczyzną pierścienia.
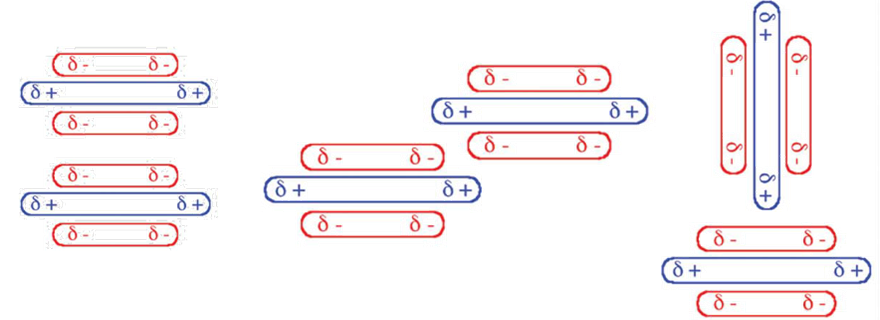
W tym przypadku wzajemne przesunięcie oddziałujących cząsteczek jest korzystne gdyż zmniejsza elektrostatyczne odpychanie pomiędzy jednoimiennymi ładunkami zlokalizowanymi w obszarach nad i pod pierścieniem, a najkorzystniejszy energetycznie jest układ w kształcie litery T. Wzajemnie równoległe ustawienie oddziałujących cząsteczek jest preferowane w przypadku gdy jeden z pierścieni aromatycznych jest destabilizowany.
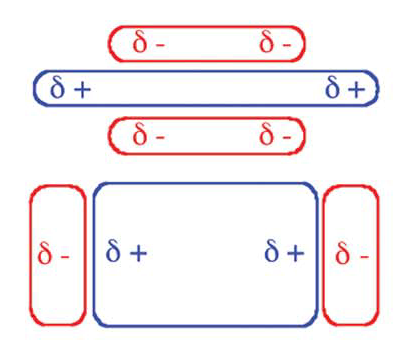
Model oddziaływania pomiędzy pierścieniami aromatycznymi, oparty na teorii orbitali cząsteczkowych został opracowany przez Huntera i Sandersa stanowi uzupełnienie modelu oddziaływań momentów kwadrupolowych cząsteczek. Zgodnie z modelem Huntera i Sandersa orbitale wiążące typu πjednego z pierścieni przekazują gęstość elektronową na antywiążące orbitale π* drugiego pierścienia aromatycznego. Dodatkowo w tym donorowo–akceptorowym wiązaniu biorą udział jednocentrowe antywiążące orbitale typu Rydbergowskiego obydwu pierścieni.
Parametry geometryczne, takie jak kąt i odległość pomiędzy pierścieniami, opisujące oddziaływanie warstwowe są uzależnione od rodzaju pierścieni jakie w nim uczestniczą. Jednak zakłada się, że w przypadku równoległego ułożenia pierścieni kąt pomiędzy nimi nie powinien być większy od 30°, a odległość pomiędzy środkami pierścieni mniejsza od 4,4 Å. Natomiast dla prostopadłego układu oddziałujących pierścieni przyjmuje się wartość kąta w granicach od 60° do 120° i odległość pomiędzy środkami pierścieni mniejszą od 5,5Å. Dla przykładu zobaczmy jak wpływa rodzaj podstawnika na energię (kcal/mol) oddziaływania pierścieni benzenowych. Weźmy pod uwagę trzy graniczne struktury:
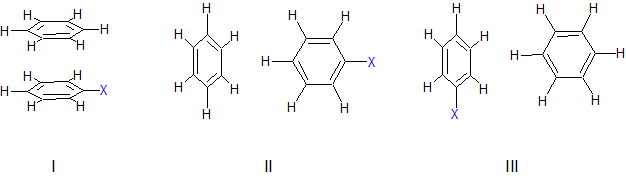
| geometria układu | |||
| X= | I | II | III |
| H | –1,8 | –2,6 | –2,6 |
| OH | –2,2 | –2,6 | –2,7 |
| CH3 | –2,2 | –2,6 | –3,0 |
| F | –2,3 | –2,8 | –2,4 |
| CN | –3,1 | –3,3 | –2,2 |
Biorąc pod uwagę powyższe dane można stwierdzić, że obecność podstawnika w pierścieniu benzenowym zwiększa siłę oddziaływania warstwowego, a ponadto zmniejsza różnice energetyczną pomiędzy formami równoległą i prostopadłą wzajemnego ustawienia oddziałujących pierścieni. W przypadku prostopadłego ustawienia pierścieni aromatycznych, forma II i III, mamy do czynienia z oddziaływaniem typu C–H...π. Obecność dowolnego podstawnika w pierścieniu benzenowym zwiększa siłę oddziaływania warstwowego gdyż zwiększa się udział sił dyspersyjnych Londona w takim układzie. W przypadku silnie destabilizujących podstawników pojawia się w układzie I oddziaływanie dipol–kwadrupol, które dodatkowo wpływa stabilizująco.
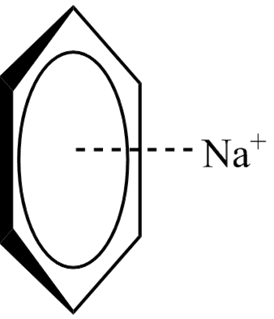 Oddziaływanie π–kation
Oddziaływanie π–kation
Oddziaływanie kation – pierścień aromatyczny jest dobrze udokumentowane, a siła tego oddziaływania zależy od rodzaju (wielkości) kationu. Przykładowe dane zostały przedstawione poniżej:
| entalpia wiązania [kcal/mol] | |
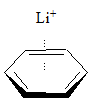 |
–39,3 |
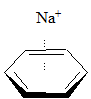 |
–28,0 |
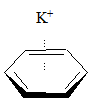 |
–19,2 |
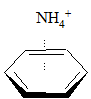 |
–19,3 |
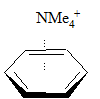 |
–9,4 |
Biorąc pod uwagę, że wartość entalpii oddziaływania jonu potasowego z wodą wynosi –17,9 kcal/mol można zauważyć, że jon ten silniej oddziałuje z pierścieniem benzenowym. Jednak efekt solwatacyjny wody jest na tyle duży, że porównywanie tylko wartości entalpii może być mylące. Chociaż w polarnych rozpuszczalnikach oddziaływania π–kation są na tyle silne, że mogą konkurować z wiązaniami wodorowymi. Przyjrzyjmy się obliczonym wartością entalpii oddziaływania czwartorzędowej soli metyloamoniowej z benzenem oraz wiązania w octanie N–metyloaminy w układach różniących się stałą dielektryczną, czyli układom:
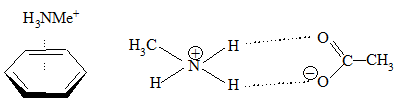
| rozpuszczalnik | stała dielektryczna | energia oddziaływania [kcal/mol] | |
| π–kation | wiązanie w soli | ||
| faza gzowa | 1,0 | –12,5 | –125,5 |
| CCl4 | 2,2 | –7,8 | –53,4 |
| octan etylu | 6,0 | –6,2 | –19,7 |
| etanol | 24,9 | –5,6 | –5,2 |
| acetonitryl | 37,5 | –5,6 | –3,8 |
| woda | 78,0 | –5,5 | –2,2 |
Jak widać rozdzielenie soli jonowej w fazie gazowej jest niekorzystne energetycznie, ale im bardziej polarny rozpuszczalnik tym, tym silniejsze staje się oddziaływanie kation–układ elektronów π. Oddziaływanie to jest związane z siłami dyspersyjnymi Londona chociaż większą rolę odgrywa tutaj elektrostatyczne przyciąganie pomiędzy cząstkowym ładunkiem ujemnym związanym z momentem kwadrupolowym benzenu i ładunkiem dodatnim kationu. Efekty związane z polaryzowalnością nie są dominujące w tego rodzaju oddziaływaniach, co można wywnioskować chociażby porównując siły oddziaływania kationu litowego z cykloheksanem i benzenem. Oszacowana moc wiązania cykloheksan–Li+ wynosi –15 kcal/mol, a benzen–Li+ około –35 kcal/mol. Natomiast energia związana z przeniesieniem ładunku z wiązania πC=C benzenu na orbitals rydbergowski 2s* Li to 3,7 kcal/mol, a wartość energii oddziaływania orbitali σC–C z 2s* Li wynosi 1,3 kcal/mol.
Optymalną geometrią dla oddziaływania kation–π jest prostopadłe ułożenie kationu nad środkiem pierścienia benzenowego, natomiast warto przeanalizować zmiany mocy tego wiązania przy zmianie geometrii układu.
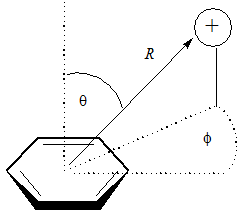 gdzie R oznacza odległość pomiędzy środkiem pierścienia a kationem; θ jest wartością kąta 90°–wartość odchylenia od płaszczyzny pierścienia, a Φ oznacza kąt rotacyjny w płaszczyźnie. W tabeli poniższej przedstawiono wybrane wartości tych wielkości i jak widać dla pewnych wartości zebranych w drugim wierszu tabeli oddziaływanie kation–π jest silne.
gdzie R oznacza odległość pomiędzy środkiem pierścienia a kationem; θ jest wartością kąta 90°–wartość odchylenia od płaszczyzny pierścienia, a Φ oznacza kąt rotacyjny w płaszczyźnie. W tabeli poniższej przedstawiono wybrane wartości tych wielkości i jak widać dla pewnych wartości zebranych w drugim wierszu tabeli oddziaływanie kation–π jest silne.
| energia oddziaływania [kcal/mol] | |||||
| θ | Φ | R [Å] | faza gazowa | chloroform | woda |
| 0 | 0 | 1,9 | –35,9 | –21,2 | –18,4 |
| 60 | 30 | 3,1 | –19,2 | –8,0 | –6,0 |
| 90 | 30 | 3,5 | –11,3 | +0,7 | –0,7 |
W przypadku gdy kation litowy znajduje się w płaszczyźnie pierścienia następuje odpychanie pomiędzy nim i pierścieniem w rozpuszczalnikach co wynika z oddziaływania dodatniego ładunku kationu z momentem kwadrupolowym pierścienia aromatycznego.
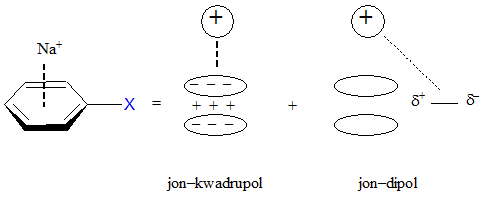 Wpływ podstawnika w pierścieniu na siłę oddziaływania π–kation
Wpływ podstawnika w pierścieniu na siłę oddziaływania π–kation
Obecność podstawników w pierścieniu aromatycznym powoduje pojawienie się czynnika odpowiedzialnego za oddziaływanie jon–dipol w całkowitej energii układu kation–π. Takie podejście nie wymaga pojawienia się znacznej polaryzacji w układzie kation–π, chociaż ma ona miejsce w przypadkach podstawników czy kationów o znacznej elektroujemności. Destabilizujące układ aromatyczny silnie akceptorowe podstawniki powodują zmniejszenie siły oddziaływania kationów z układem aromatycznym co daje widać porównując wartości energii oddziaływania kationu sodowego z pochodnymi benzenu:
| podstawnik | E [kcal/mol] |
| –N(CH3)2 | –33,9 |
| –CH3 | –28,3 |
| –H | –26,9 |
| –OH | –26,6 |
| –F | –21,8 |
| –NO2 | –14,0 |
Oddziaływanie kationów z układami heteroaromatycznymi
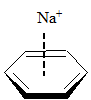
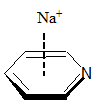
Obecność heteroatomu w pierścieniu aromatycznym powoduje dwa przeciwstawne wpływy. Z jednej strony wolne pary elektronowe heteroatomów włączane w układ sprzężonych wiązań π zwiększają siłę oddziaływania z kationem. Z drugiej strony obecność elektroujemnego atomu w pierścieniu może powodować obniżenie gęstości elektronowej zlokalizowanej w obrębie sprzężonych wiązań π i wtedy kation oddziałuje głównie z wolną parą elektronową heteroatomu. Poniżej przedstawione zostały wartości energii (kcal/mol) i momentu kwadrupolowego (a.u.) dla oddziaływania kationu sodowego z wybranymi związkami heteroaromatycznymi
 |
 |
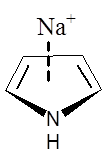 |
 |
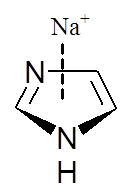 |
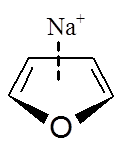 |
|
| [kcal/mol] | –27,1 | –20,0 | –29,6 | –32,6 | –21,0 | –21,0 |
| [a.u.] | –5,9 | –3,8 | –6,6 | – | –4,4 | –4,5 |
Oddziaływanie pirydyny z kationem sodowym jest słabsze niż benzenu, co wynika z destabilizującego układ wiązań podwójnych wpływu atomu azotu. Wartość momentu kwadrupolowego dla pirydyny jest mniejsza niż dla cząsteczki benzenu. Natomiast układ wiązań podwójnych w pierścieniu pirolowym jest aktywowany przez wolną parę elektronową zlokalizowaną na atomie azotu i jego oddziaływanie z kationem sodowym jest silniejsze niż w przypadku benzenu. Z drugiej strony pierścień benzenowy połączony z pirolem powoduje zwiększenie siły oddziaływania pomiędzy kationem a cząsteczką indolu, najprawdopodobniej w wyniku zwiększonego udziału sił dyspersyjnych w oddziaływaniu π–kation. W przypadku imidazolu jak i furanu następuje dezaktywacja pierścienia. Interesującym przykładem jest 1,3,5-trifluorobenzen, którego moment kwadrupolowy wynosi zero, a pomimo tego oddziaływanie cząsteczki tego związku z kationem sodowym charakteryzuje się wartością energii równą –12,4 kcal/mol. Wartość ta jest znacznie niższa niż dla układu benzen–kation sodowy, ale nie jest zerowa. Wynika to z tego, że w takim układzie rolę odgrywają oddziaływania elektrostatyczne oraz dyspersyjne i oddziaływania pomiędzy dipolami indukowanymi.