 Principles of Chemistry
Principles of Chemistry
 Wiązania kowalencyjne
Wiązania kowalencyjne
Wiązania chemiczne opisywane są dwiema teoriami. Jedna z nich teoria wiązań walencyjnych dominowała w chemii nieorganicznej przez dłuższy czas. Jednak zastosowanie modelu orbitali cząsteczkowych okazało się bardziej adekwatne (co istotne, też prostsze w interpretacji), do opisu cząsteczek związków chemicznych.
Ogólnie mówiąc dwa atomy tworzą cząsteczkę dlatego, że ich wzajemnemu zbliżeniu towarzyszy obniżenie energii całkowitej. W ogólności energię cząsteczki można uważać za sumę energii elektronów, czyli ich energię kinetyczną i potencjalną wzajemnego oddziaływania łącznie z energią potencjalną w polu jąder atomowych traktowanych jako spoczywające w położeniu równowagi, wzajemnej energii potencjalnej jąder oraz energii oscylacyjnej, rotacyjnej ii translacyjnej cząsteczki jako całości. Te trzy ostatnie rodzaje energii, czyli oscylacyjna, rotacyjna i translacyjna stanowią niewielki ułamek całkowitej energii cząsteczki. Z tego powodu pojęcie całkowitej energii cząsteczki jest praktycznie równoważne energii elektronowej uzupełnionej przez energię kulombowskiego odpychania jąder. Oddziaływania typu grawitacyjnego pomiędzy jądrami można pominąć. Odsunięciu dwóch jąder wodoru od siebie do nieskończoności biorąc pod uwagę jedynie przyciąganie grawitacyjne wymaga energii równej 2,5·10–54 J podczas gdy rzeczywista praca równa jest 6,7·10-19 J, czyli jest przeszło 1035 razy większa.
Posługując się pojęciem całkowitej energii elektronowej czyli energii elektronów łącznie z kulombowską energią odpychania jąder, które przyjmujemy jako nieruchome, możemy stwierdzić, że teoria wiązania chemicznego musi wyjaśniać i przewidywać jak energia elektronowa zależy od położenia jąder, przewidzieć konfigurację elektronową cząsteczki i wskazać jak zmienia się energia gdy cząsteczka ulega odkształceniom. Założenie dotyczące nieruchomości jąder powoduje, że rozpatrywana w ten sposób elektronowa energia wiązania nie jest identyczna z tą jaka wyznaczamy doświadczalnie czyli energią dysocjacji. Różnice wynikają z trzech powodów. Po pierwsze należy uwzględnić zerową energię oscylacji, która w przypadku dużych cząsteczek wieloatomowych może osiągać wartości porównywalne z energią wymaganą do rozerwania jakiegokolwiek pojedynczego wiązania. Chociaż dla cząsteczek dwuatomowych jej udział w całkowitej energii cząsteczki mieści się w przedziale od 0.1 do 0.05. Następny czynnik to energia translacyjna równa 3/2KT obejmująca translacje całej cząsteczki jak i wszystkich jej fragmentów z osobna. Trzecim czynnikiem jest energia rotacyjna cząsteczki jako całości, która w temperaturach bliski 0 K wynosi 3/2KT w przypadku cząsteczki nieliniowej i KT dla cząsteczki liniowej.
Zachodzące w cząsteczce zmiany energii często przedstawia się w postaci wykresu energii potencjalnej. Zgodnie z wykresem gdy odległość pomiędzy atomami t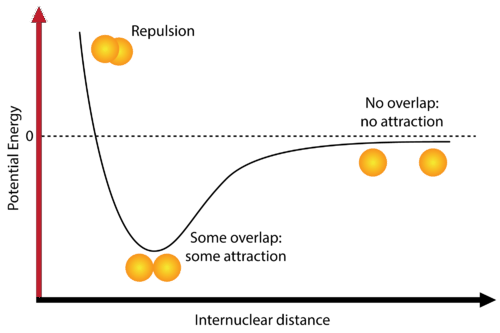 worzącymi cząsteczkę jest duża to całkowita energia elektronowa jest zbliżona do sumy energii elektronowych izolowanych atomów tworzących cząsteczkę. Gdy atomy zbliżają się do siebie to energia cząsteczki osiąga minimum przy pewnej wartości odległości pomiędzy jądrami odpowiadającej stanowi równowagi jąder w trwałej cząsteczce. Przy odległościach mniejszych od wartości równowagowej krzywa wznosi się gdyż wiążące oddziaływanie elektronów traci na znaczeniu, a dominujące staje się wzajemne odpychanie między jądrami atomów. Jednym z problemów teorii wiązania chemicznego jest dokładny opis krzywej energii potencjalnej cząsteczki. Dobrym przybliżeniem przebiegu krzywej energii potencjalnej jest empiryczny wzór Morse’a z roku 1920, który przyjmując, że energia odpowiadająca nieskończenie oddalonym od siebie jądrom jest równa zero czyli E(R)=EAB(R) – (EA-EB), jest następujący:
worzącymi cząsteczkę jest duża to całkowita energia elektronowa jest zbliżona do sumy energii elektronowych izolowanych atomów tworzących cząsteczkę. Gdy atomy zbliżają się do siebie to energia cząsteczki osiąga minimum przy pewnej wartości odległości pomiędzy jądrami odpowiadającej stanowi równowagi jąder w trwałej cząsteczce. Przy odległościach mniejszych od wartości równowagowej krzywa wznosi się gdyż wiążące oddziaływanie elektronów traci na znaczeniu, a dominujące staje się wzajemne odpychanie między jądrami atomów. Jednym z problemów teorii wiązania chemicznego jest dokładny opis krzywej energii potencjalnej cząsteczki. Dobrym przybliżeniem przebiegu krzywej energii potencjalnej jest empiryczny wzór Morse’a z roku 1920, który przyjmując, że energia odpowiadająca nieskończenie oddalonym od siebie jądrom jest równa zero czyli E(R)=EAB(R) – (EA-EB), jest następujący:
E(R) =De[exp{–2a(R–Re)} – 2exp{–a(R–Re)}],
gdzie a jest stałą; De równowagową odległością pomiędzy jądrami cząsteczki. W przypadku cząsteczki wieloatomowej zamiast krzywej energii wyznacza się powierzchnię energii potencjalnej gdyż energia takiej cząsteczki zależy od kilku zmiennych niezależnych takich jak długości wiązań i kąty między tymi wiązaniami.
Opis wiązań w cząsteczkach związków chemicznych opiera się pojęciach i metodzie mechaniki kwantowej. Próbując rozwiązać równanie falowe dla cząsteczki wieloelektronowej, nawet przyjmując założenie o nieruchomości jąder, musimy poczynić dalsze uproszczenia. Wprowadzono dwa podejścia do tego problemu. Jedno oparte na pierwszych rachunkach Heitlera i Londona z roku 1927 dotyczących cząsteczki wodoru, i drugie oparte o rozbudowę powłok atomowych, w którym elektronom przypisuje się niezależne, własne orbitale. Pierwsze ujęcie nosi nazwę teorii wiązań walencyjnych – VB – od angielskich słów valence bond. Drugie podejście to teoria orbitali cząsteczkowych – MO – molecular orbitals. W teorii VB konstrukcja funkcji falowej podkreśla rolę oddzielnych atomów i zlokalizowanych orbitali atomowych. W metodzie orbitali cząsteczkowych elektronom przypisuje się orbitale zajmujące całą cząsteczkę, co kładzie nacisk na delokalizację elektronów i ich uwspólnienie. W teorii obydwie metody powinny dawać takie same wyniki, jednak najczęściej obliczenia prowadzone są w pierwszym przybliżeniu i wtedy należy mieć świadomość, którą z metod wykorzystano do obliczeń. Początkowo metoda wiązań walencyjnych była wykorzystywana szerzej, jednak obecnie obliczenia prowadzi się w prostszej matematycznie metodzie orbitali cząsteczkowych.
Charakter wiązania kowalencyjnego, jak i każdego innego rodzaju, uzależniony jest od takich czynników jak ładunek jąder atomowych tworzących cząsteczkę, konfiguracji elektronowej pierwiastków, rozmiarów atomów i innych. Te czynniki są charakterystyczne dla każdego pierwiastka, a w związku z tym należy oczekiwać zróżnicowania w obrębie wiązań kowalencyjnych. Oczywistym jest, że wiązania w cząsteczkach heterojądrowych będą spolaryzowane gdyż chmura elektronowa tworząca wiązanie będzie silniej przyciągana przez atom pierwiastka o większej elektroujemności. W cząsteczkach homojądrowych mamy do czynienia z "czystym" wiązaniem kowalencyjnym. Dokładne omówienie wiązań w teoriach VB i MO znajduje się w części Atom i cząsteczka, tutaj natomiast skupimy się na opisie ilościowym kowalencyjności wiązania.
Polarność wiązania przejawia się doświadczalnie jako elektryczny moment dipolowy cząsteczki, co w teorii orbitali cząsteczkowych odpowiada nierówności współczynników obu orbitali atomowych tworzących wiązanie (orbital wiążący). Inaczej mówiąc dla cząsteczki dwuatomowej A i B wiązanie opisujemy jako:
ψ = N(φA + λφB).
Przy granicznych wartościach współczynnika λ, czyli λ→0 lub λ→∞ obydwa elektrony wiązania skupiałyby się albo przy atomie A lub atomie B. W efekcie powstawałoby wiązanie czysto jonowe opisywane jako A–B+, lub A+B–. Natomiast gdy parametr λ≈1 nie zauważymy żadnego przesunięcia elektronów i wiązanie będzie czysto kowalencyjne.
Przemieszczenie ładunku, związane z różnicą elektroujemności atomów tworzących cząsteczkę i wiązanie dwuelektronowe, w teorii orbitali cząsteczkowych można opisać równaniem:
P = 2φ2 = 2N2[φ2A + 2λφAφB + λ2φ2B]
Czynnik normalizujący N jest równy ∫P dτ=2 gdyż ilość ładunku odpowiada dwóm elektronom. Ponieważ: ∫φ2A dτ = ∫φ2B dτ = 1 i ∫φAφB dτ = S, czyli całce nakrywania, to podstawiając do wzoru te wartości otrzymujemy 2=2N2[1 + 2λS + λ2], a stąd już łatwo wyznaczyć wartość czynnika normalizującego:
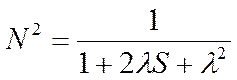
Wzór na przemieszczenie ładunku P wygodnie jest zapisać w postaci::
P= PAAφ2A + PBBφ2B + 2PABφAφB
gdzie populacje elektronowe wynoszą odpowiednio PAA= qA, PBB= qB i 2PAB=qABS., a suma ładunków cząstkowych qA+ qB. + qAB=2. W takim wypadku populacje orbitali i obszaru nakrywania są równe:
qA= 2N2, qB= 2λ2N2 i qAB= 4λN2S. Teraz wzór na P uzyskuje postać:
P = qAdA + qBdB + qABdAB
gdzie dA = φ2A, dB = φ2B, dAB = φAB/S, czyli mamy dwie gęstości orbitalne i jedną gęstość nakrywania. Teraz jeżeli λ≠1 i qA ≠ qB to przemieszczenie łądunku nadaje wiązaniu charakter jonowy. Za miarę charakteru jonowego wiązania – FIC (fractional ionic character) – przyjęto za Klesingerem i McWeeny wartość
FIC= (qA + qB)/2, który w teorii orbitali cząsteczkowych jest równy:
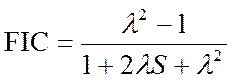 Wartość graniczna równa 0 ,tego wyrażenia, odpowiada wiązaniu kowalencyjnemu (λ=1), natomiast równa 1 – wiązaniu jonowemu (λ→∞).
Wartość graniczna równa 0 ,tego wyrażenia, odpowiada wiązaniu kowalencyjnemu (λ=1), natomiast równa 1 – wiązaniu jonowemu (λ→∞).
Ponieważ przeniesienie ładunku przejawia się powstaniem momentu dipolowego, a ilość ładunku na jednostkę objętości jest równa –eP, to:
![]() Moment dipolowy stanowi sumę części elektronowej, pochodzącej od elektronów walencyjnych i elektrycznego momentu dipolowego pozostałych jonów dodatnich , z których każdy utracił jeden elektron oddając go wiązaniu. Przyjmując, że te ostatnie przyczynki pochodzą od ładunków punktowych wypadkowy moment dipolowy można powiązać ze współrzędnymi z i członami elektronowymi qi równaniem:
Moment dipolowy stanowi sumę części elektronowej, pochodzącej od elektronów walencyjnych i elektrycznego momentu dipolowego pozostałych jonów dodatnich , z których każdy utracił jeden elektron oddając go wiązaniu. Przyjmując, że te ostatnie przyczynki pochodzą od ładunków punktowych wypadkowy moment dipolowy można powiązać ze współrzędnymi z i członami elektronowymi qi równaniem:
![]()
Aby powiązać tak opisany moment dipolowy z parametrem FIC musimy założyć, że orbitale φA i φB są zwykłymi, (nie zhybrydyzowanymi, orbitalami atomowymi, a odległości z odpowiadają połowie odległości pomiędzy środkami ciężkości ładunków w cząsteczce. Centroid gęstości nakrywania φAφB pokrywa się ze środkiem wiązania pomiędzy atomami A i B. Przy takich założeniach moment dipolowy wyraża równanie:
μ = –eR•FIC.
Natomiast powiązanie momentu dipolowego z parametrem λ i całką nakrywania wyraża się zależnością:
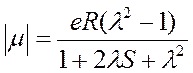 W metodzie wiązań walencyjnych konieczne jest dołączenie członów jonowych do funkcji falowej, a wyrażenia opisujące gęstości ładunków qA i qB stają się bardziej skomplikowane. Ostatecznie wzory na FIC i moment dipolowy przyjmują postać:
W metodzie wiązań walencyjnych konieczne jest dołączenie członów jonowych do funkcji falowej, a wyrażenia opisujące gęstości ładunków qA i qB stają się bardziej skomplikowane. Ostatecznie wzory na FIC i moment dipolowy przyjmują postać:
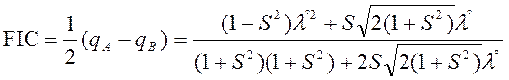 i
i
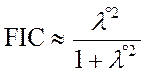 Przy czym parametr λ° oznacza wagę pewnej jonowej struktury w dwuelektronowej funkcji falowej.
Przy czym parametr λ° oznacza wagę pewnej jonowej struktury w dwuelektronowej funkcji falowej.
Opis wiązania za pomocą orbitali atomowych, jak to czynimy w teorii MO nie do końca odpowiada rzeczywistości. Dopiero wprowadzenie pewnego udziału hybrydyzacji pozwala na uzyskanie wartości teoretycznych dobrze odpowiadających danym eksperymentalnym. Uwzględnienie hybrydyzacji powoduje powstanie dipola atomowego gdyż pierwotnie centrosymetryczne orbitale atomowe ulegają deformacji. W przypadku cząsteczek heterojądrowych centroid nakładania orbitali φAφB jest przesunięty w stosunku do środka ciężkości, co oznacza, że gęstość nakrywania ma udział w momencie dipolowym. Taki sam efekt występuje nawet w przypadku cząsteczki, w której wiązanie jest tworzone przez orbitale o różnych rozmiarach. Dodatkowym czynnikiem, który należy mieć na uwadze jest obecność innych poza pojedynczymi, typu σ, wiązaniami w cząsteczce. Na koniec należy wspomnieć o udziale elektronów nie biorących udziału w tworzeniu wiązania, które mogą wnosić wkład w powstanie momentu dipolowego. Szczególnie jest to istotne w przypadku obecności wolnych par elektronowych, które w przeciwieństwie do kulistosymetrycznych orbitali atomowych są silnie odkształcone, a przez to wnoszą istotny wkład do momentu dipolowego cząsteczki.