 Principles of Chemistry
Principles of Chemistry
 Związki koordynacyjne z ligandami π–akceptorowymi
Związki koordynacyjne z ligandami π–akceptorowymi
Zgodnie ze swoją nazwą ligandy π–akceptorowe są cząsteczkami lub jonami posiadającymi akceptorowe orbitale o symetrii π. W związku koordynacyjnym zawierającym tego typu ligandy rząd wiązania M–L jest większy od jedności. Biorąc pod uwagę, że wiązanie metal–ligand w takich układach odbywa się za pomocą donorowych elektronów d atomu centralnego i akceptorowego orbitalu liganda wiązania te określa się nazwą odwrotnej koordynacji. Związki koordynacyjne z tego typu ligandami odgrywają ważną rolę w chemii koordynacyjnej czy to ze względów teoretycznych gdyż są to związki charakteryzujące się bardzo wysoką kowalencyjnością wiązań, czy też pod względem praktycznym. Wiele związków tego typu jest dobrymi katalizatorami szeregu reakcji chemicznych. Należy podkreślić, że ligandy π–akceptorowe stabilizują niskie stopnie utlenienia jonów centralnych.
Karbonylki
Pierwsze doniesienia o reakcji tlenku węgla z metalami pochodzą z roku 1834, kiedy to Justus von Liebig badał reakcję zachodzącą podczas przepuszczania tlenku węgla nad stopionym potasem. Reakcja ta nie prowadzi do powstania karbonylku potasu, ale skład otrzymanego produktu Liebig określił na KCO. Pierwszy karbonylek metalu – [Pt(CO)2Cl2] – został otrzymany przez Paula Schützenbergera w roku 1868 w wyniku przepuszczania mieszaniny chloru i tlenku węgla nad czernią platynową. W latach 90-tych dziewiętnastego stulecia Ludwig Mond wraz z Carlen Langerem i Friedrichem Quincke prowadzili badania nad odzyskiwanie chloru z odpadów powstających w metodzie Solvaya. Między innymi próbowali do tego celu wykorzystać nikiel i zaobserwowali powstawanie łatwo rozkładającego się związku niklu, który po podgrzaniu do 100°C rozkładał się na nikiel i tlenek węgla. Mond i współpracownicy odkryli tym samym tetrakarbonylek niklu o wzorze [Ni(CO)4] będą cy bezbarwną cieczą o temperaturze wrzenia 43°C.W następnym roku Mond i niezależnie od niego Pierre Eugène Marcellin Berthelot odkryli pentakarbonylek żelaza. Opierając się na badaniach Monda, i korzystając z jego pomocy finansowej, Heinrich Hirtz i M. Dalton Cowap opracowali metody syntezy karbonylków kobaltu, molidbenu, rutenu oraz [Fe2(CO)9]. W roku 1927 A. Job i A. Cassal otrzymali heksakarbonylki chromu i wolframu. W 1930 roku Walter Hieber usystematyzował metody otrzymywania karbonylków metali, a opierając się na opracowanej przez siebie metodzie otrzymywania uzyskał karbonylek renu – [Re2(CO)10].
Metody syntezy
Niektóre karbonylki metali przejściowych można otrzymać w wyniku bezpośredniej reakcji pomiędzy metalem i tlenkiem węgla. Przykładem może być karbonylek niklu(0), który powstaje już w temperaturze 80°C pod ciśnieniem atmosferycznym. Synteza pentakarbonyleku żelaza(0) wymaga zastosowania temperatur w zakresie od 150°C do 200°C i ciśnienia CO w zakresie 5•106 do 2•107 Pa. Karbonylki wanadu, chromu, wolframu, molibdenu otrzymuje się przez redukcję chlorków tych metali reduktorami jak glin, etyloglin, glinowodorek litu, lub też wykorzystuje się tlenek węgla jako reduktor, jak na przykład w reakcji otrzymywania dekakarbonylku direnu(0)
Re2O7 + 17CO → [Re2(CO)10] + 7CO2
Rodzaje koordynacji CO
Związki koordynacyjne zawierające ligandy karbonylowe możemy podzielić na dwa główne rodzaje:
- homoleptyczne – zawierające w sferze koordynacyjnej tylko ligandy CO
- heteroleptyczne – w sferze koordynacyjnej oprócz CO znajdują się też inne ligandy.
Biorąc pod uwagę liczbę atomów centralnych karbonylki, podobnie jak i inne związki koordynacyjne, możemy podzielić na jednordzeniowe i wielordzeniowe. W wielordzeniowych karbonylkach bardzo często występują wiązania metal–metal. Dodatkowo w przypadku takich związków możemy wyróżnić te, które zawierają tylko jeden rodzaj liganda (CO) i noszą one ogólną nazwę związków izoleptycznych.
Ligand karbonylowy może koordynować do metalu różnorako, od prostej koordynacji terminalnej do różnorodnych typów koordynacji mostkowej.
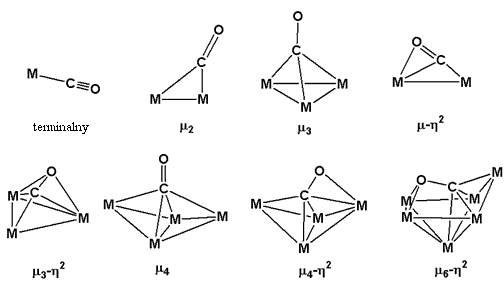 Wielościany koordynacyjne jednordzeniowych karbonylków odpowiadają ich liczbom koordynacyjnym, czyli heksakarbonylki charakteryzują się oktaedrycznym wielościanem koordynacyjnym (Oh), Symetrię D3h wykazują pentakarbonylki, Td tetrakarbonylki.
Wielościany koordynacyjne jednordzeniowych karbonylków odpowiadają ich liczbom koordynacyjnym, czyli heksakarbonylki charakteryzują się oktaedrycznym wielościanem koordynacyjnym (Oh), Symetrię D3h wykazują pentakarbonylki, Td tetrakarbonylki.
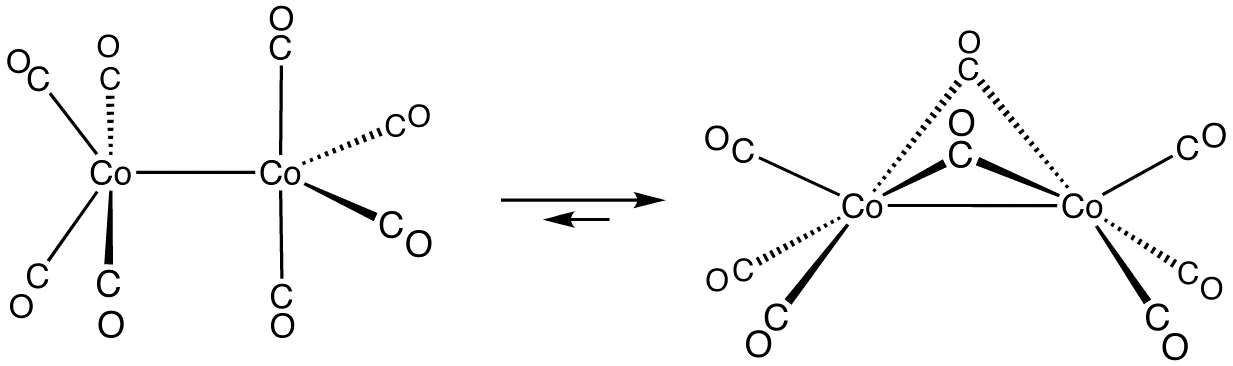
W przypadku wielordzeniowych karbonylków mogą występować mostowe grupy CO, lub też tworzyć się wiązania metal–metal. Karbonylek [Co2(CO)8] należy jednocześnie do obydwu rodzajów. W roztworze występuje w postaci dimeru bez mostkowych grup CO, a w ciele stałym dwie grupy karbonylowe tworzą mostki:
Spektroskopia IR
Widma w podczerwieni karbonylków metali dostarczają istotnych informacji o strukturze tych związków. Gazowy tlenek węgla charakteryzuje się pasmem absorpcyjnym νCO z maksimum przy 2143 cm−1. Energia pasma νCO dla skoordynowanego tlenku węgla zmienia się w zależności od siły oddziaływania zwrotnego, a tym samym od rzędu wiązania C–O. Przykładowe wartości podane zostały w poniższej tabeli:
| Związek | νCO [cm−1] |
| CO | 2143 |
| [Ti(CO)6−2] | 1748 |
| [V(CO)6−1] | 1859 |
| [Cr(CO)6] | 2000 |
| [Mn(CO)5+] | 2100 |
| [Fe(CO)42+] | 2204 |
| [Fe(CO)5] | 2022, 2000 |
Ilość pasm, na widmach IR, odpowiadających grupie karbonylowej jest uzależniona od geometrii wielościanu koordynacyjnego i ilości grup karbonylowych skoordynowanych do atomu centralnego. Dodatkowo jeszcze należy mieć na uwadze, że przedstawione w poniższej tabeli liczby pasm absorpcyjnych dotyczą pomiarów wykonanych w roztworach lub fazie gazowej. W ciele stałym ilości pasm absorpcyjnych mogą być większe co wynika z symetrii otoczenia cząsteczki związku.

Wiązanie metal–CO
Tlenek węgla nie wykazuje typowych właściwości kwasowo-zasadowych, nie tworzy połączeń z BF3 czy metalami alkalicznymi jak sód czy potas. Jednocześnie istnieje liczna grupa jego związków z metalami d<-elektronowymi.
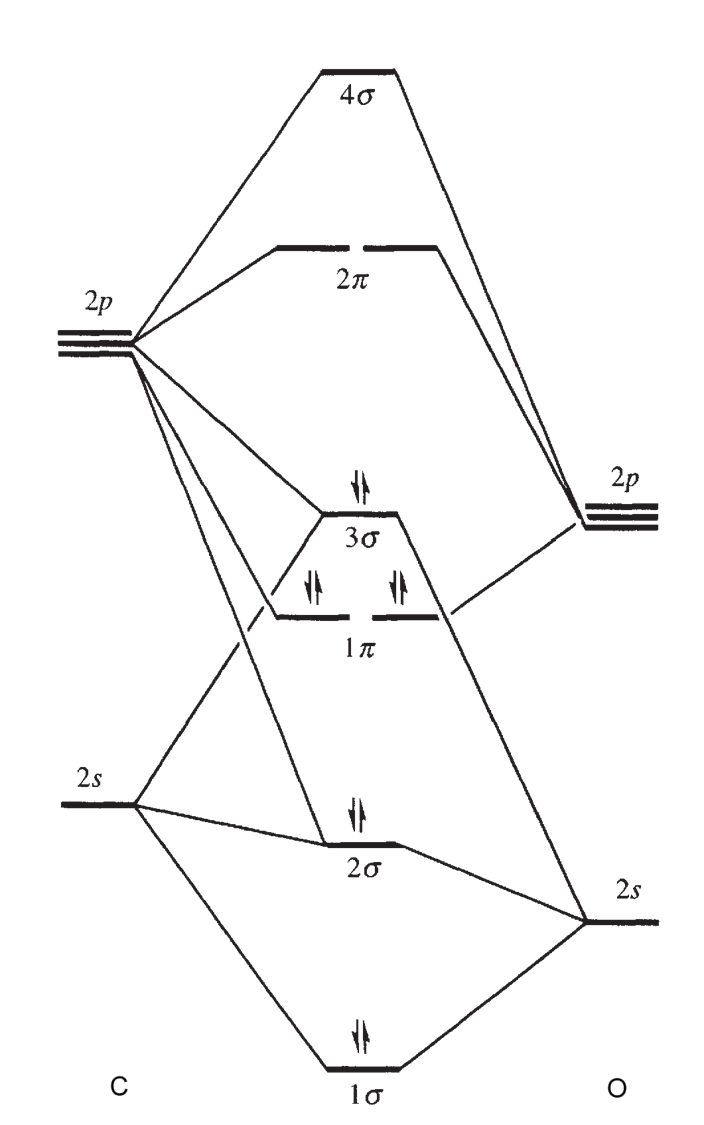
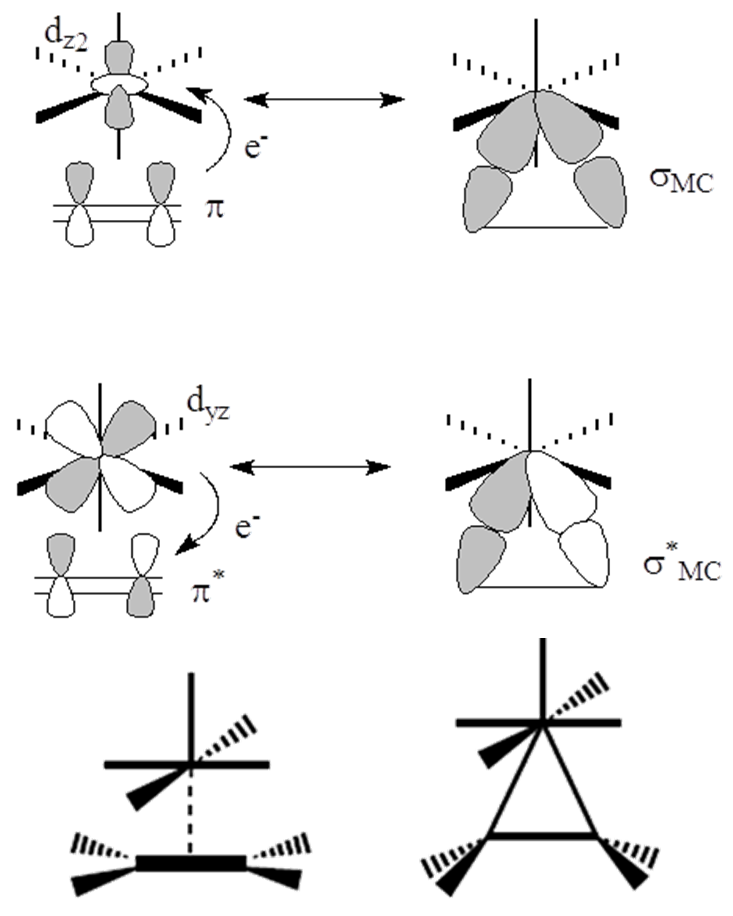
Rozważmy strukturę elektronową cząsteczki CO. Orbital HOMO jest zlokalizowany na atomie węgla i na nim umieszczona jest para elektronowa 3σ. Gęstość elektronowa na orbitalu 1π (HOMO-1) jest przesunięta w kierunku atomu tlenu ze względu na jego większą elektroujemność. Związki karbonylowe są charakterystyczne dla metali na niskich stopniach utlenienia co wskazuje na zaangażowanie w tworzenie wiązań metal-karbonyl antywiążących „pustych” czyli akceptorowych orbitali 1π*, zlokalizowanych na atomie węgla Zgodnie z tym możemy przedstawić za Dewarem-Chattem-Duncansonen tworzenie wiązania metal-karbonyl w sposób następujący:
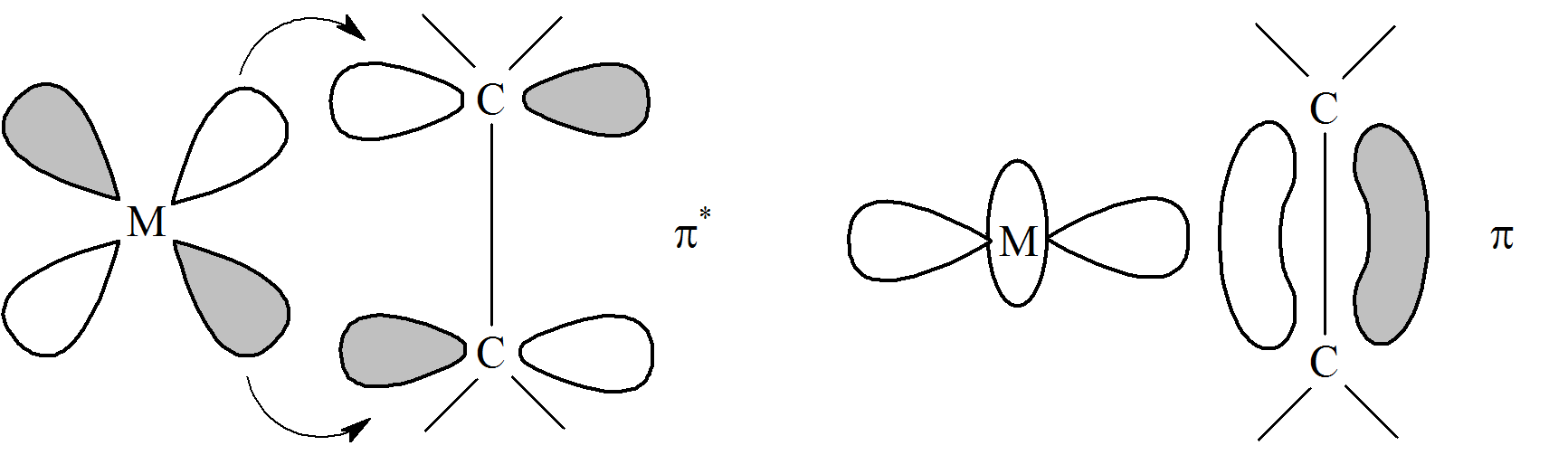
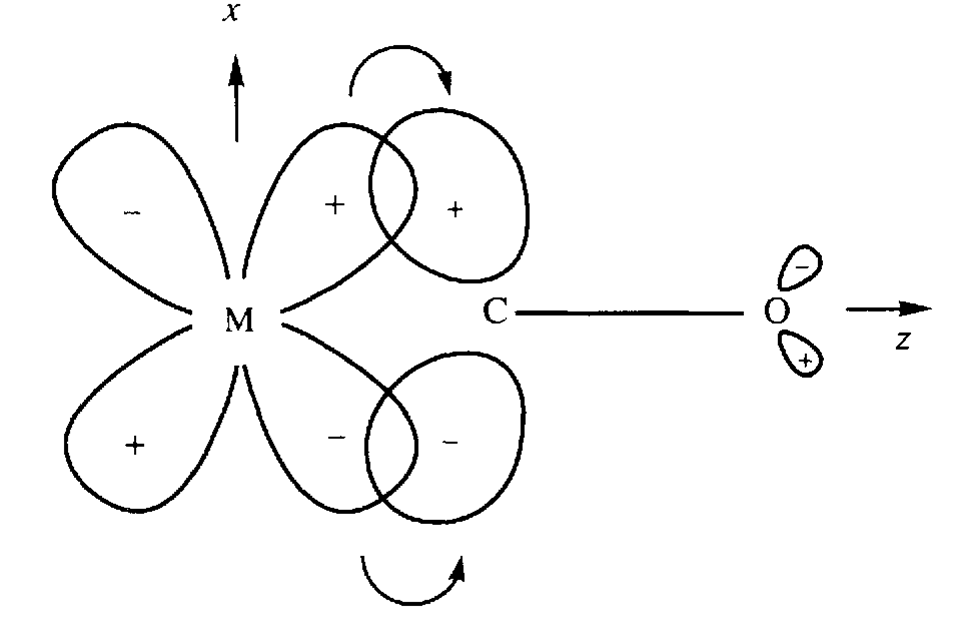 Jak widać gęstość elektronowa zlokalizowana na orbitalach dπ atomu centralnego (dxz, dxy) jest przenoszona na antywiążący orbital 1π* tlenku węgla. W ten sposób cząsteczka CO uzyskuje ładunek ujemny. Tworzeniu wiązania s towarzyszy proces związany z odwrotnym przemieszczeniem ładunku z orbitalu 3σ tlenku węgla na akceptorowy orbital dz2 jonu centralnego.
Jak widać gęstość elektronowa zlokalizowana na orbitalach dπ atomu centralnego (dxz, dxy) jest przenoszona na antywiążący orbital 1π* tlenku węgla. W ten sposób cząsteczka CO uzyskuje ładunek ujemny. Tworzeniu wiązania s towarzyszy proces związany z odwrotnym przemieszczeniem ładunku z orbitalu 3σ tlenku węgla na akceptorowy orbital dz2 jonu centralnego.
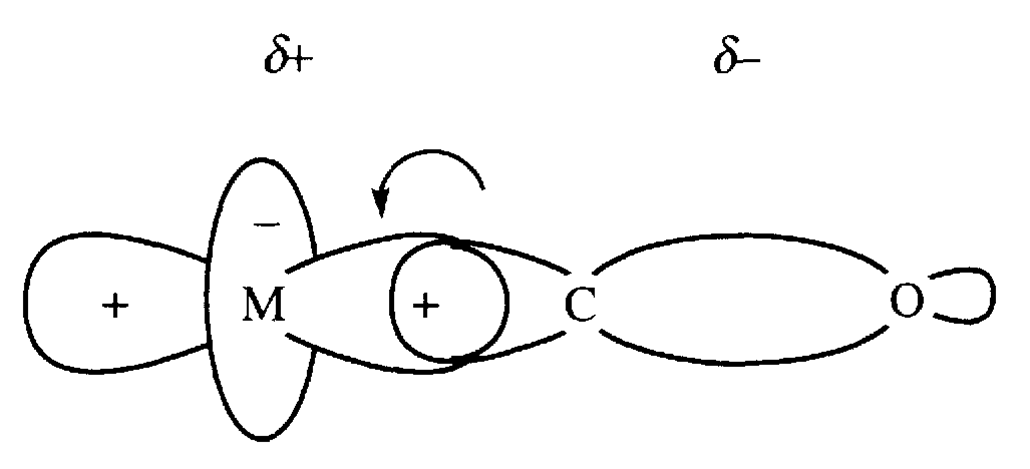 Utworzenie wiązania metal – tlenek węgla w karbonylku metalu jest procesem, w którym przeniesienie gęstości elektronowej ze strony metalu do CO jest skorelowane z odwrotnym przekazaniem gęstości elektronowej przy tworzeniu wiązania σ. Własności σ-donorowe grupy karbonylowej są wzmacniane przez kwasowość CO wynikającą z własności orbitali 1π*. Takie podejście do tworzenia wiązania metal-CO wyjaśnia dlaczego karbonylki są tworzone przez jony centralne na niskich stopniach utlenienia. Gdy metal nie posiada elektronów, które mogą podlegać przeniesieniu na akceptorowe orbitale tlenku węgla, nie jest możliwe utworzenie wiązania π, a tym samym synergia z wiązaniem σ.
Utworzenie wiązania metal – tlenek węgla w karbonylku metalu jest procesem, w którym przeniesienie gęstości elektronowej ze strony metalu do CO jest skorelowane z odwrotnym przekazaniem gęstości elektronowej przy tworzeniu wiązania σ. Własności σ-donorowe grupy karbonylowej są wzmacniane przez kwasowość CO wynikającą z własności orbitali 1π*. Takie podejście do tworzenia wiązania metal-CO wyjaśnia dlaczego karbonylki są tworzone przez jony centralne na niskich stopniach utlenienia. Gdy metal nie posiada elektronów, które mogą podlegać przeniesieniu na akceptorowe orbitale tlenku węgla, nie jest możliwe utworzenie wiązania π, a tym samym synergia z wiązaniem σ.
W oktaedrycznych karbonylkach orbitale dxz i dyz atomu centralnego mogą oddziaływać z wiążącym i antywiążącym orbitalem π tlenku węgla (1π i 2π); odpowiednio πx i π*x z dxy oraz πz i π*z z dyz jak przedstawiono na schemacie:
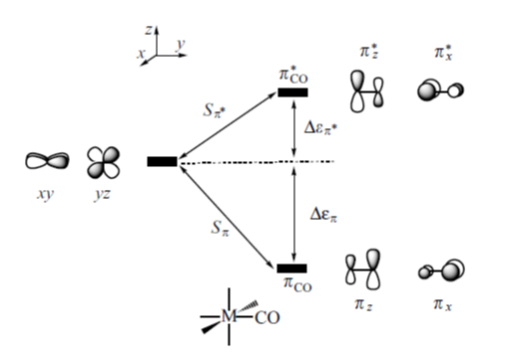 Porównanie siły oddziaływania d → π oraz d → π* powinno umożliwić określenie czy ligand karbonylowy w danym związku koordynacyjnym zachowuje się jako π-donor czy π-akceptor. Ponieważ wzajemne oddziaływania metal–CO są synergiczne to całkowita siła wiązania jest związana jednocześnie z obniżeniem energii orbitali i zwiększeniem różnicy energetycznej pomiędzy poziomem wiążącym a antywiążącym. Jednak samo porównanie wartości Dεπ i Dεπ* nie jest jednoznaczne gdyż energia orbitali d zależy od rodzaju metalu. Biorąc pod uwagę tylko kryterium energetyczne, oddziaływanie atomu centralnego z antywiążącymi orbitalami π* tlenku węgla jest silniejsze niż d → π dla metali przejściowych od skandu do manganu co sugeruje, że w tych karbonylkach ligand zachowuje się jako π-akceptor. Jednak w przypadku takich metali jak Co, Ni oraz Cu obniżenie energii poziomu πCO jest stosunkowo małe. Uwzględnienie wielkości energii pomiędzy orbitalami wiążącym i antywiążącym CO oraz uwzględnienie polaryzacji orbitali πCO i π*CO, wpływającej na ich nakładanie się z orbitalami d atomu centralnego pozwala stwierdzić że, preferencje π-akceptorowe liganda karbonylowego występują dla karbonylków metali w szeregu Sc-Mn, a preferencje π-donorowe dla metali od żelaza do miedzi. W przypadku karbonylków miedzi obydwa czynniki czyli nakładanie orbitali metalu z orbitalami tlenku węgla (d → π*) oraz czynnik energetyczny (d → π) posiadają bardzo zbliżony wkład w tworzenie wiązania metal-karbonyl. Podobna sytuacja, to znaczy występowanie niskoenergetycznych orbitali d atomu centralnego, jest charakterystyczna dla metali przejściowych czwartego i następnych okresów. Takie przedstawienie wiązania M–CO wymaga jego rozpatrzenia jako układu obejmującego orbitale d atomu metalu i jednocześnie orbitale wiążące i antywiążące π tlenku węgla. Bardzo często ligand karbonylowy jest traktowany w sposób uproszczony przez uwzględnienie jedynie antywiążących orbitali π*CO. W takim ujęciu para elektronowa zlokalizowana na atomie węgla odpowiada za σ-donorowe właściwości cząsteczki CO, a dwa puste orbitale π*są związane z silnymi elektronoakceptorowymi właściwościami liganda karbonylowego. Takie przedstawienie wiązania M-CO wymaga jego rozpatrzenia jako układu obejmującego orbitale d atomu metalu i jednocześnie orbitale wiążące i antywiążące π tlenku węgla. Bardzo często ligand karbonylowy jest traktowany w sposób uproszczony przez uwzględnienie jedynie antywiążących orbitali π*CO. W takim ujęciu para elektronowa zlokalizowana na atomie węgla odpowiada za σ-donorowe właściwości cząsteczki CO, a dwa puste orbitale π* są związane z silnymi elektronoakceptorowymi właściwościami liganda karbonylowego.
Porównanie siły oddziaływania d → π oraz d → π* powinno umożliwić określenie czy ligand karbonylowy w danym związku koordynacyjnym zachowuje się jako π-donor czy π-akceptor. Ponieważ wzajemne oddziaływania metal–CO są synergiczne to całkowita siła wiązania jest związana jednocześnie z obniżeniem energii orbitali i zwiększeniem różnicy energetycznej pomiędzy poziomem wiążącym a antywiążącym. Jednak samo porównanie wartości Dεπ i Dεπ* nie jest jednoznaczne gdyż energia orbitali d zależy od rodzaju metalu. Biorąc pod uwagę tylko kryterium energetyczne, oddziaływanie atomu centralnego z antywiążącymi orbitalami π* tlenku węgla jest silniejsze niż d → π dla metali przejściowych od skandu do manganu co sugeruje, że w tych karbonylkach ligand zachowuje się jako π-akceptor. Jednak w przypadku takich metali jak Co, Ni oraz Cu obniżenie energii poziomu πCO jest stosunkowo małe. Uwzględnienie wielkości energii pomiędzy orbitalami wiążącym i antywiążącym CO oraz uwzględnienie polaryzacji orbitali πCO i π*CO, wpływającej na ich nakładanie się z orbitalami d atomu centralnego pozwala stwierdzić że, preferencje π-akceptorowe liganda karbonylowego występują dla karbonylków metali w szeregu Sc-Mn, a preferencje π-donorowe dla metali od żelaza do miedzi. W przypadku karbonylków miedzi obydwa czynniki czyli nakładanie orbitali metalu z orbitalami tlenku węgla (d → π*) oraz czynnik energetyczny (d → π) posiadają bardzo zbliżony wkład w tworzenie wiązania metal-karbonyl. Podobna sytuacja, to znaczy występowanie niskoenergetycznych orbitali d atomu centralnego, jest charakterystyczna dla metali przejściowych czwartego i następnych okresów. Takie przedstawienie wiązania M–CO wymaga jego rozpatrzenia jako układu obejmującego orbitale d atomu metalu i jednocześnie orbitale wiążące i antywiążące π tlenku węgla. Bardzo często ligand karbonylowy jest traktowany w sposób uproszczony przez uwzględnienie jedynie antywiążących orbitali π*CO. W takim ujęciu para elektronowa zlokalizowana na atomie węgla odpowiada za σ-donorowe właściwości cząsteczki CO, a dwa puste orbitale π*są związane z silnymi elektronoakceptorowymi właściwościami liganda karbonylowego. Takie przedstawienie wiązania M-CO wymaga jego rozpatrzenia jako układu obejmującego orbitale d atomu metalu i jednocześnie orbitale wiążące i antywiążące π tlenku węgla. Bardzo często ligand karbonylowy jest traktowany w sposób uproszczony przez uwzględnienie jedynie antywiążących orbitali π*CO. W takim ujęciu para elektronowa zlokalizowana na atomie węgla odpowiada za σ-donorowe właściwości cząsteczki CO, a dwa puste orbitale π* są związane z silnymi elektronoakceptorowymi właściwościami liganda karbonylowego.
Nitrozylowe związki koordynacyjne
Związki nitrozylowe zawierają w sferze koordynacyjnej ligand NO. Jeżeli przyjmiemy, że ligand nitrozylowy występuje w tego typu związkach w postaci kationu NO+, to biorąc pod uwagę izoelektronowość NO+ i tlenku węgla możemy rozpatrywać wiązania w cząsteczkach związków nitrozylowych w analogiczny sposób jak w karbonylkach.

Ligand nitrozylowy jest donorem dwóch elektronów, a poprzez oddziaływanie synergiczne akceptuje elektrony od metalu. Skoordynowany ligand NO może przybierać dwie formy, liniową i zgiętą.
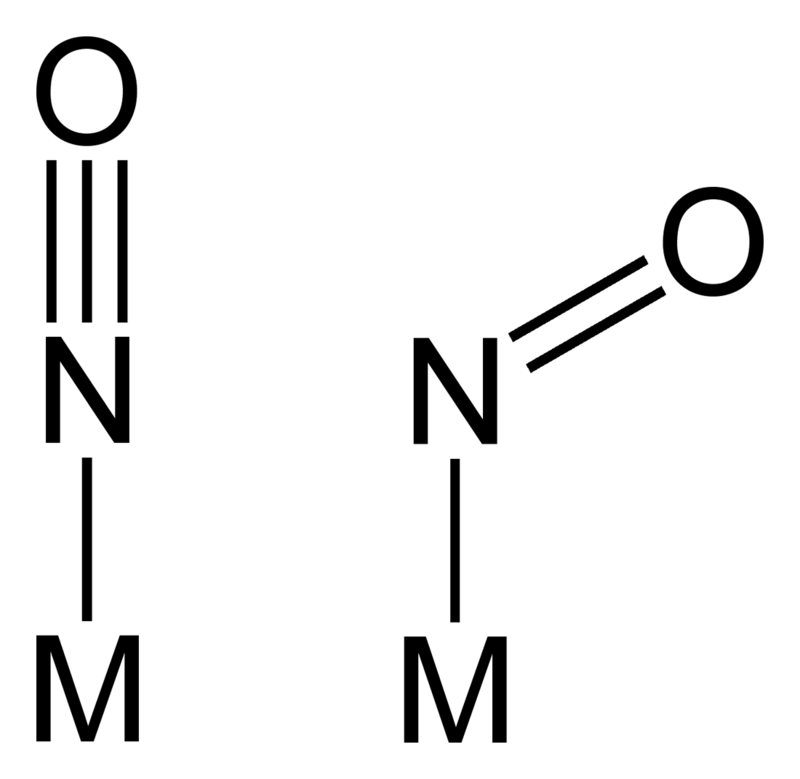 W formie liniowej jest on opisywany jako anion NO–. geometria liganda nitrozylowego jest uzależniona od siły oddziaływania zwrotnego w związku koordynacyjnym. Jeżeli atom centralny zawiera większą od 6 liczbę elektronów π, ligand NO przyjmuje w związkach o oktaedrycznej geometrii wielościanu koordynacyjnego formę zgiętą. Geometrię liganda nitrozylowego można określić na podstawie położenia pasm w zakresie podczerwieni. Liniowa geometria układu M–NO absorbuje w zakresie 1525–1690 cm−1, podczas gdy zgięta forma grupy nitrozylowej charakteryzuje się pasma mi absorpcyjnymi w zakresie 1650–1900 cm−1, co ma związek z krotnością wiązań w ligandzie nitrozylowym.
W formie liniowej jest on opisywany jako anion NO–. geometria liganda nitrozylowego jest uzależniona od siły oddziaływania zwrotnego w związku koordynacyjnym. Jeżeli atom centralny zawiera większą od 6 liczbę elektronów π, ligand NO przyjmuje w związkach o oktaedrycznej geometrii wielościanu koordynacyjnego formę zgiętą. Geometrię liganda nitrozylowego można określić na podstawie położenia pasm w zakresie podczerwieni. Liniowa geometria układu M–NO absorbuje w zakresie 1525–1690 cm−1, podczas gdy zgięta forma grupy nitrozylowej charakteryzuje się pasma mi absorpcyjnymi w zakresie 1650–1900 cm−1, co ma związek z krotnością wiązań w ligandzie nitrozylowym.
Etylenowe związki koordynacyjne
Zbliżony model tworzenia wiązań występuje w przypadku związków koordynacyjnych z ligandem etylenowym, na przykład soli Zeisa K[PtCl3(C2H4)]. Tutaj też następuje oddziaływanie pomiędzy zajętymi orbitalami typu p cząsteczki etylenu i akceptorowymi orbitalami d metalu połączone z przesunięciem gęstości elektronowej od metalu na antywiążące, akceptorowe orbitale π* C2H4.
 W przypadku związków koordynacyjnych z ligandem etylenowym występuje pewna nieokreśloność charakteru związku. Otóż, przesunięcie ładunku orbitalu π układu C=C na akceptorowy orbital dz2 metalu powoduje obniżenie gęstości elektronowej wiązania węgiel-węgiel, osłabienie wiązania i konsekwentnie wzrost jego długości. Ponadto oddziaływanie zwrotne dyz → π* zwiększa gęstość elektronową na antywiążącym orbitalu π alkenu co również osłabia wiązanie C=C. Dodatkowo obydwa te oddziaływania mają charakter wiążący. Pojawia się w związku z tym dylemat czy alken występuje tutaj jako cząsteczkowy etylen, czy też należy ten układ rozpatrywać jako cyklometaloetan z wiązaniem pojedynczym węgiel-węgiel i podwójnym wiązaniem metal-węgiel. Należy zatem rozważyć formy mezomeryczne układu. Omawiany proces możemy przedstawić następującym schematem:
W przypadku związków koordynacyjnych z ligandem etylenowym występuje pewna nieokreśloność charakteru związku. Otóż, przesunięcie ładunku orbitalu π układu C=C na akceptorowy orbital dz2 metalu powoduje obniżenie gęstości elektronowej wiązania węgiel-węgiel, osłabienie wiązania i konsekwentnie wzrost jego długości. Ponadto oddziaływanie zwrotne dyz → π* zwiększa gęstość elektronową na antywiążącym orbitalu π alkenu co również osłabia wiązanie C=C. Dodatkowo obydwa te oddziaływania mają charakter wiążący. Pojawia się w związku z tym dylemat czy alken występuje tutaj jako cząsteczkowy etylen, czy też należy ten układ rozpatrywać jako cyklometaloetan z wiązaniem pojedynczym węgiel-węgiel i podwójnym wiązaniem metal-węgiel. Należy zatem rozważyć formy mezomeryczne układu. Omawiany proces możemy przedstawić następującym schematem:
 Opis stopnia utlenienia jonu centralnego w układzie metal-etylen jest też niejednoznaczny, gdyż w przypadku formy cyklicznej stopnień utlenienia metalu jest wyższy od 2. Badania strukturalne związków koordynacyjnych z ligandem etylenowym również nie prowadzą do jednoznacznego określenia, która z form mezomerycznych jest dominująca. Długości wiązań C–C w tych związkach wynoszą około 1,43 Å i mają pośrednią wartość pomiędzy długością wiązania pojedynczego (1,54 Å) a podwójnego (1,32 Å). Również geometria układu M–CH2 nie jest płaska, lecz piramidalna. Dodatkowym czynnikiem, który ma wpływ na geometrię układu są inne ligandy występujące w cząsteczce związku. Im silniejsze jest oddziaływanie pomiędzy jonem centralnym a układem etylenowym tym struktura związku bardziej zbliża się do metalocyklopropanu, i jednocześnie im słabsze oddziaływanie metal–etylen tym bardziej układ przypomina formę mezomeryczną, w której ligand etylenowy występuje jako cząsteczkowy C2H4. Innymi słowy układ metal-etylen mogą opisywać obydwie przedstawione powyżej formy.
Opis stopnia utlenienia jonu centralnego w układzie metal-etylen jest też niejednoznaczny, gdyż w przypadku formy cyklicznej stopnień utlenienia metalu jest wyższy od 2. Badania strukturalne związków koordynacyjnych z ligandem etylenowym również nie prowadzą do jednoznacznego określenia, która z form mezomerycznych jest dominująca. Długości wiązań C–C w tych związkach wynoszą około 1,43 Å i mają pośrednią wartość pomiędzy długością wiązania pojedynczego (1,54 Å) a podwójnego (1,32 Å). Również geometria układu M–CH2 nie jest płaska, lecz piramidalna. Dodatkowym czynnikiem, który ma wpływ na geometrię układu są inne ligandy występujące w cząsteczce związku. Im silniejsze jest oddziaływanie pomiędzy jonem centralnym a układem etylenowym tym struktura związku bardziej zbliża się do metalocyklopropanu, i jednocześnie im słabsze oddziaływanie metal–etylen tym bardziej układ przypomina formę mezomeryczną, w której ligand etylenowy występuje jako cząsteczkowy C2H4. Innymi słowy układ metal-etylen mogą opisywać obydwie przedstawione powyżej formy.
Związki z cząsteczkowym azotem jako ligandem
Azot cząsteczkowy, co wynika z jego budowy elektronowej, jest bierny chemicznie. jedynym metalem reagującym z azotem jest lit, ale w wyniku reakcji tworzą się azotki, w których wiązanie w cząsteczce N2 ulega zerwaniu. Cząsteczka dizotu jest izoelektronowa z cząsteczką tlenku węgla, ale nie tworzy analogów heksakarbonylków. Natomiast znane są związki niektórych metali przejściowych zawierające w sferze koordynacyjnej cząsteczki N2. Co odróżnia je od karbonylków to fakt, że w sferze koordynacyjnej metalu poza ligandem diazotkowym występują ligandy fosfinowe. Pośród możliwych typów koordynacji N2 eksperymentalnie potwierdzono cztery rodzaje:
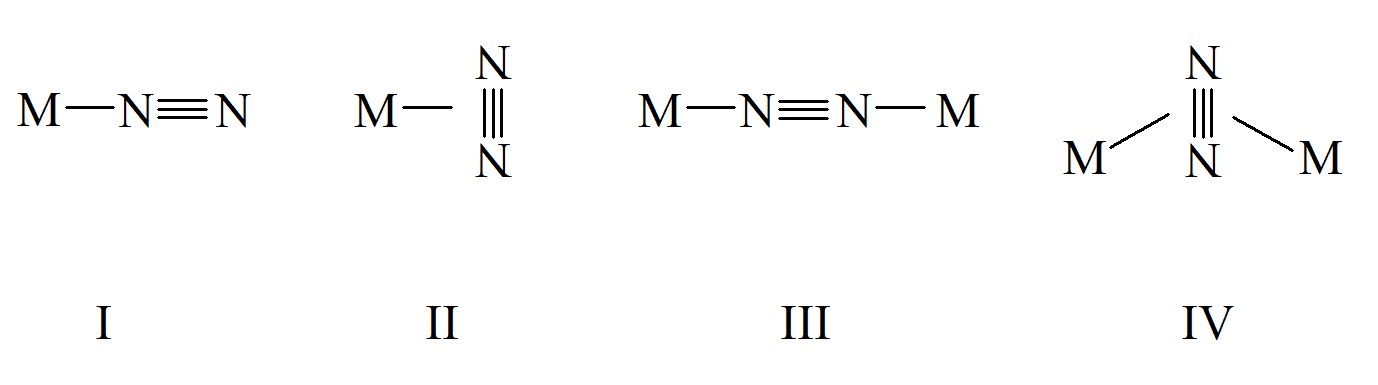 Większość związków koordynacyjnych ligandem diazotowym ma budowę liniową I i III, z katami wiązań M–N≡N bliskimi 180°. Wiązania pomiędzy cząsteczką N2 a atomem metalu odbywa się z udziałem donorowych elektronów diazotu i akceptorowych orbitali atomu centralnego (dz2, dx2–y2). Oczywiście ma tutaj tez miejsce oddziaływanie zwrotne pomiędzy donorowymi orbitalami dπ metalu i jednym z akceptorowych orbitali antywiążących π* N2. Rząd wiązania M–N2 jest, w większości tego typu związków, większy od jedności. W wyniku przeniesienia gęstości elektronowej od metalu do cząsteczki diazotu rząd wiązania, a tym samym i odległość pomiędzy atomami azotu ulega zwiększeniu w porównaniu do swobodnej cząsteczki N2, chociaż zmiany długości wiązania N–N nie są duże. Osłabienie siły wiązania w skoordynowanej cząsteczce diazotu przejawia się przesunięciem częstości drgań walencyjnych νN–N z 2331 cm-1 dla diazotu do wartości od 1800 do 2100 cm-1. Można oczekiwać, że w przypadku dwurdzeniowych związków koordynacyjnych, w których cząsteczka diazotu jest koordynowana do dwóch atomów centralnych, wydłużenie wiązania N–N będzie większe niż w przypadku jednordzeniowych związków. Jednak długość wiązania azot – azot w skoordynowanej terminalnie cząsteczce diazotu jest tylko nieznacznie krótsze od wartości jakie występują w przypadku koordynacji mostkowej. Z drugiej strony wydłużenie wiązania jest uzależnione od rodzaju ligandów występujących w sferze koordynacyjnej i od metalu centralnego, do którego koordynowany jest diazot.
Większość związków koordynacyjnych ligandem diazotowym ma budowę liniową I i III, z katami wiązań M–N≡N bliskimi 180°. Wiązania pomiędzy cząsteczką N2 a atomem metalu odbywa się z udziałem donorowych elektronów diazotu i akceptorowych orbitali atomu centralnego (dz2, dx2–y2). Oczywiście ma tutaj tez miejsce oddziaływanie zwrotne pomiędzy donorowymi orbitalami dπ metalu i jednym z akceptorowych orbitali antywiążących π* N2. Rząd wiązania M–N2 jest, w większości tego typu związków, większy od jedności. W wyniku przeniesienia gęstości elektronowej od metalu do cząsteczki diazotu rząd wiązania, a tym samym i odległość pomiędzy atomami azotu ulega zwiększeniu w porównaniu do swobodnej cząsteczki N2, chociaż zmiany długości wiązania N–N nie są duże. Osłabienie siły wiązania w skoordynowanej cząsteczce diazotu przejawia się przesunięciem częstości drgań walencyjnych νN–N z 2331 cm-1 dla diazotu do wartości od 1800 do 2100 cm-1. Można oczekiwać, że w przypadku dwurdzeniowych związków koordynacyjnych, w których cząsteczka diazotu jest koordynowana do dwóch atomów centralnych, wydłużenie wiązania N–N będzie większe niż w przypadku jednordzeniowych związków. Jednak długość wiązania azot – azot w skoordynowanej terminalnie cząsteczce diazotu jest tylko nieznacznie krótsze od wartości jakie występują w przypadku koordynacji mostkowej. Z drugiej strony wydłużenie wiązania jest uzależnione od rodzaju ligandów występujących w sferze koordynacyjnej i od metalu centralnego, do którego koordynowany jest diazot.
 Wiązanie metal–diazot w dwurdzeniowych związkach koordynacyjnych
Wiązanie metal–diazot w dwurdzeniowych związkach koordynacyjnych
 Wiązanie π metal-diazot w przypadku koordynacji liniowej i bocznej cząsteczki diazotu.
Wiązanie π metal-diazot w przypadku koordynacji liniowej i bocznej cząsteczki diazotu.
Utworzenie wiązań π pomiędzy metalem i skoordynowaną cząsteczką diazotu powoduje przeniesienie gęstości elektronowej na N2 a tym samym wytworzenie efektywnego, ujemnego, ładunku na tym ligandzie. Z tego powodu diazotowe związki koordynacyjne zachowują się jak zasady Lewisa. Ligand diazotowy w takich związkach może ulegać protonowaniu, co często prowadzi do eliminacji tego liganda. Fosfinowe związki koordynacyjne wolframu(0) zawierające w sferze koordynacyjnej dwie cząsteczki diazotu reagują z roztworem kwasu siarkowego(VI) w roztworze metanolowym, w temperaturze pokojowej, z wydzieleniem amoniaku. Czynnikiem redukującym w takim układzie jest atom wolframu, który utlenia się do +6 stopnia.