 Principles of Chemistry
Principles of Chemistry
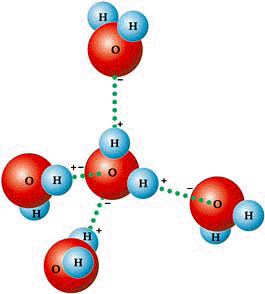 Wiązania wodorowe
Wiązania wodorowe
To słabe wiązanie chemiczne polegające głównie na elektrostatycznym oddziaływaniu pomiędzy atomem wodoru i elektroujemny atomem posiadającym wolne pary elektronowe jest fundamentalnym oddziaływaniem w organizacji organizmów żywych stanowiąc podstawę większości procesów biologicznych i biochemicznych. Wiązania wodorowe są odpowiedzialne za asocjację cząsteczek wody, tworzenie podwójnej helisy DNA, a tym samym za jej właściwości związane z przechowywaniem oraz
przekazywaniem informacji genetycznych podczas replikacji, a także za drugorzędową strukturę białek i agregację enzymów.
Koncepcja wiązania wodorowego pojawiła się z początkiem XX wieku ale sam termin został zdefiniowany dopiero w roku 1920 w pracy Latimera i Rodebusha zatytułowanej „A definitive discussion of the H bond”. Wiązanie wodorowe jest specyficznym oddziaływaniem bliskiego zasięgu, w którym cząsteczka kwasu Brönsteda, protonodonora R–X–H, oddziałuje z cząsteczką zasady Y–R, tak zwanym – centrum protonoakceptorowym. W wyniku tego procesu powstaje układ, w którym pozbawiony elektronu atom wodoru znajduje się w obszarze o dużej gęstości elektronowej.
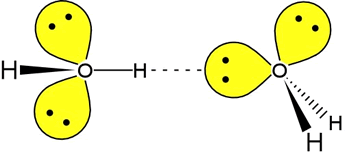 Aby mogło powstać wiązanie wodorowe konieczne jest, aby atomy X i Y miały małe promienie kowalencyjne oraz silnie elektroujemny charakter w porównaniu z atomem wodoru. W związku z tym protonoakceptorami są atomy posiadające wolne pary elektronowe, np.: O, S, N, chlorowce i ich jony, ale mogą być to też być grupy atomów powiązane elektronami π, czyli wiązania wielokrotne lub układy aromatyczne. Donorami protonu w mostkach wodorowych X–H···Y są natomiast grupy: –OH, –NH, –SH, –CH, –XH (X – atom chlorowca).
Aby mogło powstać wiązanie wodorowe konieczne jest, aby atomy X i Y miały małe promienie kowalencyjne oraz silnie elektroujemny charakter w porównaniu z atomem wodoru. W związku z tym protonoakceptorami są atomy posiadające wolne pary elektronowe, np.: O, S, N, chlorowce i ich jony, ale mogą być to też być grupy atomów powiązane elektronami π, czyli wiązania wielokrotne lub układy aromatyczne. Donorami protonu w mostkach wodorowych X–H···Y są natomiast grupy: –OH, –NH, –SH, –CH, –XH (X – atom chlorowca).
Właściwości fizykochemiczne związków polarnych takich jak: alkohole, aldehydy, kwasy karboksylowe czy też amidy, są w znacznej mierze wynikiem istnienia wiązań wodorowych za pomocą których cząsteczki tych związków łączą się w większe zespoły – asocjaty. Oddziaływania te wymuszają określoną orientację molekuł i prowadzą do zwiększenia sztywności takiego układu cząsteczek, w porównaniu z układami cząsteczek niezasocjowanych. Postaci tworzących się asocjatów zależą przede wszystkim od struktury cząsteczek, gdyż może występować tzw. efekt steryczny, który utrudnia zbliżenie molekuł na taką odległość, aby możliwe było utworzenie wiązania wodorowego.
Klasyfikacja wiązań wodorowych
Położenie protonu w mostku wodorowym opisują krzywe energii potencjalnej, których wykresy można przedstawić następująco:
 Teraz w zależności od rodzaju atomów wchodzących w skład mostka wodorowego oraz siły tworzonego przez atomy wiązania wodorowego możemy wyróżnić dwa typy wiązań wodorowych:
Teraz w zależności od rodzaju atomów wchodzących w skład mostka wodorowego oraz siły tworzonego przez atomy wiązania wodorowego możemy wyróżnić dwa typy wiązań wodorowych:
– asymetryczne wiązanie wodorowe – z tym rodzajem wiązania wodorowego mamy do czynienia, gdy atomy X i Y tworzące mostek wodorowy (X–H···Y) posiadają różną elektroujemność. Krzywa energii potencjalnej jest niesymetryczna i posiada dwa minima o różnej głębokości (a). Proton znajduje się w głębiej leżącym minimum, a po doprowadzeniu energii aktywacji przeskakuje do płytszego minimum. Prawdopodobieństwo takiego przejścia uzależnione jest od wysokości bariery potencjału oraz od temperatury, która decyduje o obsadzeniu poziomów oscylacyjnych w cząsteczce. Krzywa energii potencjalnej staje się coraz bardziej symetryczna (b), a bariera potencjału rozdzielająca minima coraz węższa i niższa, w wyniku zmiany siły wiązania wodorowego. Pociąga to za sobą zmianę długości omawianego wiązania. Stąd najdłuższemu wiązaniu odpowiada krzywa przedstawiona na rysunku (a), gdzie proton znajduje się przy donorze, natomiast najkrótsze wiązanie wodorowe przedstawia krzywa zaprezentowana na rysunku (d).
– symetryczne wiązanie wodorowe – występuje pomiędzy dwoma identycznymi atomami o tej samej elektroujemności (X–H···X). Krzywa energii potencjalnej jest symetryczna (c) i ma dwa równoważne minima dla protonu, które są rozdzielone przez płaskie maksimum. Proton zlokalizowany jest w jednym położeniu, aczkolwiek w wyniku doprowadzenia energii może pokonać barierę potencjału i przeskoczyć do sąsiedniego minimum. Zmniejszanie się odległości międzyjądrowej powoduje wzrost siły wiązania i przejście w krzywą z pojedynczym, szerokim minimum (d).
W zależności od liczby akceptorów z jaką oddziałuje grupa protonodonorowa X–H w wiązaniu wodorowym możemy wyróżnić:
– wiązanie dwucentrowe (liniowe), gdzie atom wodoru jest połączony z dwoma różnymi atomami tj. X–H···Y. Tego rodzaju wiązania występują najczęściej w przypadku silnych wiązań wodorowych;
– wiązanie trzycentrowe, w którym atom wodoru łączy się z trzema różnymi atomami, z jednym przez wiązanie kowalencyjne, a z dwoma pozostałymi przez wiązania wodorowe, przy czym można wyróżnić tutaj wiązania dwuakceptorowe X–H···(Y1,Y2) lub dwudonorowe (X1–H, X2–H)···Y. Trzycentrowe wiązania wodorowe istnieją w układach posiadających wiązania wodorowe o średniej mocy. Występują dość często w cząsteczkach węglowodanów, nukleozydów, nukleotydów oraz białek;
– wiązanie czterocentrowe, X–H···(Y1, Y2, Y3), występuje niezwykle rzadko w strukturach krystalograficznych (<5%). W przypadku tego rodzaju wiązań wodorowych kąt pomiędzy atomami X–H···Y musi być większy niż 90°. Odległość X–H jest większa niż dla wiązań trzycentrowych, stąd te wiązania są uważane czasem jako oddziaływania niewiążące.
Najczęściej stosowanym podziałem jest oparty na energii wiązania, zgodnie z którym wiązania wodorowe dzieli się na
– średniej mocy, jeśli jego energia odpowiada energii wiązań wodorowych istniejących pomiędzy cząsteczkami wody czy też cząsteczkami węglowodorów, tj. 4-15 kcal/mol,
– silne, o mocy większej niż 15 kcal/mol,
– słabe a o energii mniejszej od 4 kcal/mol.
| X–H....Y | silne wiązanie wodorowe | wiązanie wodorowe o średniej mocy | słabe wiązanie wodorowe |
| rodzaj oddziaływań | silnie kowalencyjne | głównie elektrostatyczne | elektrostatyczne/ dyspersyjne |
| długość wiązania | H...Y ~ X–H | H...Y > X–H | H...Y >> X–H |
| wydłużenie wiązania X–H [Å] | 0,05 – 0.2 | 0,01 – 0.05 | ≤0,01 |
| długość wiązania X...Y [Å] | 2,2 – 2,5 | 2,2 – 3,2 | 3,2 – 4,0 |
| długość wiązania H...Y [Å] | 1,2 – 1,5 | 1,5 – 2,2 | 2,2 – 3,0 |
| względne przesunięcie częstości pasma w IR [cm-1] | > 25% | 5 – 25% | < 5% |
| przesunięcie chemiczne w 1H NMR | 14 – 22 | < 14 | – |
| przykłady |
[F…H…F]– [N…H…N]+ P–OH…O=P |
O–H…O=C N–H…O=C O–H…O–H |
C–H…O O–H…p Os–H…O |
Kolejny podział opiera się na kryterium typowości grup protonodonorowych i protonoakceptorowych. Zgodnie z tym dzieli się wiązania wodorowe na:
– konwencjonalne – czyli takie, w których atom wodoru związany chemicznie z atomem pierwiastka silnie elektroujemnego X (tj. O, S, N czy też halogenu) przyciąga siłami elektrostatycznymi elektrony należące do silnie elektroujemnego atomu Y. Własnością klasycznych wiązań wodorowych jest tzw. „red shift”, czyli przesunięcie częstości drgania walencyjnego X–H w kierunku niższych wartości, wynikające z wydłużenia i osłabienia tego wiązania.
| bardzo silne | silne | słabe |
| [F–H…F]– |
N–H…O=C |
O–H…O O–H…S S–H…O O–H…F–C O–H…Cl–C |
– niekonwencjonalne wiązania wodorowe – termin ten odnosi się do wiązań wodorowych, które pozornie przypominają klasyczne wiązania wodorowe. Można je przedstawić formalnie jako X–H···Y, jednakże wykazują zachowanie przeciwne w stosunku do typowych wiązań wodorowych. Wiązania tego typu nie spełniają kryteriów spektroskopowych i energetycznych, gdyż cechą charakterystyczną dla tego typu wiązań wodorowych jest tzw. „blue shift”, czyli przesunięcie częstości drgania walencyjnego X–H w kierunku wyższych wartości, skorelowane ze skróceniem tego wiązania oraz obniżeniem intensywności pasma dla drgania walencyjnego X–H. Najczęściej wiązaniami niekonwencjonalnymi są słabe wiązania wodorowe.
| bardzo silne | silne | słabe |
| – |
N+–H···π O–H···π N–H···N–B X–H···C |
C–H···O C–H···N O/N–H···π C–H···π O–H···M M–H···O P–H···O O/N–H···P O/N–H···Se C–H···F–C Si–H···O |
Niekonwencjonalne wiązania wodorowe są odpowiedzialne za tworzenie struktur „nadcząsteczkowych”, istotnych z punktu widzenia chemii supramolekularnej. Tego typu wiązania wodorowe umożliwiają powstawanie zarówno naturalnych struktur supramolekularnych, takich jak helisa DNA, błony komórkowe czy kompleksy enzymów z koenzymami, ale również tworzenie struktur nieistniejących w przyrodzie, takich jak nanorurki, ciekłe kryształy, dendrymery, etery koronowe i wiele innych.
Energia wiązania wodorowego
Początkowo wiązanie wodorowe rozważano jako oddziaływanie o charakterze czysto elektrostatycznym. Takie podejście było zgodne z faktem, że atom wodoru posiadający jedynie orbital 1s może tworzyć tylko jedno wiązanie kowalencyjne. Rozwój technik badawczych, a zwłaszcza dane spektroskopowe pokazały, że obok oddziaływań elektrostatycznych, ważną rolę w tworzeniu wiązania wodorowego odgrywają również oddziaływania wywołane przeniesieniem ładunku pomiędzy orbitalami (charge-transfer). W roku 1957 Coulson zaproponował podział energii wiązania wodorowego na pięć składowych: energię oddziaływania elektrostatycznego, energię polaryzacyjną, energię dyspersji, energię przeniesienia ładunku
i energię odpychania. Przy czym do energii wiązania w silnych wiązaniach wodorowych największy wkład ma energia oddziaływań elektrostatycznych. W przypadku wiązań wodorowych o małej mocy decydującą rolę pełni energia charge-transfer. Coulson oszacował, że oddziaływanie elektrostatyczne stanowi 65% całkowitej energii oddziaływań cząsteczkowych w wiązaniu wodorowym typu O–H···O, tak więc odgrywa ono główną rolę w stabilizacji wiązania wodorowego. Kolejny model wiązania powstała w latach siedemdziesiątych dwudziestego wieku, a został opracowany przez Morokumę i współpracowników. Według tego modelu energia oddziaływania międzycząsteczkowego wyraża się wzorem:
ΔE = ES + PL + EX + CT + MIX,
gdzie
ES – oddziaływanie elektrostatyczne, tj. oddziaływanie pomiędzy niezaburzonym rozkładem ładunku elektronowego monomeru A i monomeru B. Ten człon można także rozszerzyć na wszelkie oddziaływania wyższego rzędu np. dipol-dipolowe czy też dipolowokwadrupolowe, może mieć on charakter przyciągający lub odpychający;
PL – oddziaływanie polaryzacyjne, o charakterze przyciągającym. Człon ten wyraża efekt polaryzacji ładunku elektronowego monomeru A przez monomer B (i odwrotnie) oraz oddziaływania wyższego rzędu.
EX – energia wymiany; odnosi się do oddziaływań o charakterze odpychającym, spowodowanych nakładaniem się chmur elektronowych monomeru A i B.
CT – energia przeniesienia ładunku (charge-transfer) wyraża oddziaływanie wywołane przez wymianę ładunku pomiędzy zajętym orbitalem molekularnym monomeru A i wolnym orbitalem molekularnym monomeru B (a także odwrotnie) oraz oddziaływaniami wyższego rzędu.
MIX – człon sprzęgający, wyrażający różnicę pomiędzy całkowitą energią oddziaływań wiązań wodorowych ΔE a sumą czterech wyżej wymienione wkładów, uwzględniający oddziaływania wyższego rzędu pomiędzy składowymi energii.
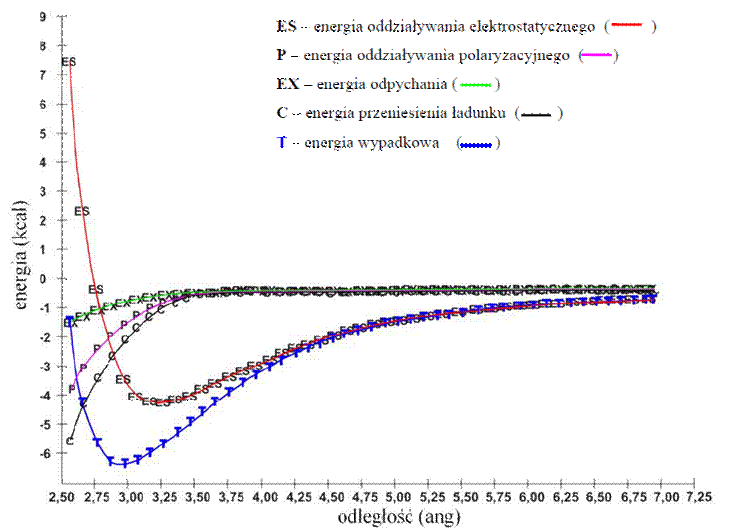 Model Morokumy powstał na gruncie teorii wiązań walencyjnych i miał zastosowanie do układów wiązań wodorowych posiadających nie więcej niż kilka elektronów.
Model Morokumy powstał na gruncie teorii wiązań walencyjnych i miał zastosowanie do układów wiązań wodorowych posiadających nie więcej niż kilka elektronów.
Oddziaływania kooperatywne w układach wiązań wodorowych
W teorii wiązań walencyjnych, na której oparł się Morokuma jak i w teorii orbitali cząsteczkowych występuje koncepcja energii całkowitej, oddziałujących ze sobą pojedynczych lub wielokrotnych wiązań kowalencyjnych. Energia całkowita takiego układu jest inna niż suma energii poszczególnych składników. W teorii wiązań walencyjnych tę dodatkową energię definiuje się jako energię rezonansową, natomiast w teorii orbitali
cząsteczkowych ta „nadmiarowa” energia określana jest jako energia delokalizacji.
Oznaczając pojedyncze cząsteczki kolejnymi literami alfabetu A, B, C, itd., całkowitą energię układu wiązań wodorowych można przedstawić następującym równaniem:
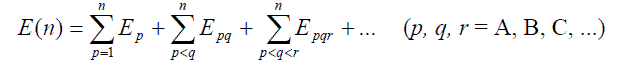
gdzie: n – liczba wiązań wodorowych w układzie,
Ep – energia pojedynczych izolowanych cząsteczek,
Epq– energia oddziaływań kooperatywnych dwóch sąsiadujących cząsteczek,
Epqr – energia oddziaływań kooperatywnych trzech cząsteczek, itd.
Różnica pomiędzy wartością energii całkowitej asocjatów wiązań wodorowych i wartością sumy energii monomerycznych wiązań wodorowych jest związana przede wszystkim z polaryzowalnością. Ze względu na wzajemną polaryzację grup funkcyjnych tworzących wiązania wodorowe rozróżnia się dwa rodzaje kooperatywności:
– kooperatywność wiązań σ – przepływ ładunku następuje wzdłuż wiązań σ grup funkcyjnych X–H (np. O–H, N–H, S–H) powodując zwiększenie polarności tych grup. Ten typ nieaddytywności wiązań wodorowych występuje w układach zawierających grupy funkcyjne, które mają jednocześnie własności protonodonorowe i protonoakceptorowe (np. O–H) i jest obserwowany m.in. w strukturze krystalicznej lodu, hydratach, węglowodanach i cyklodekstrynach. Obliczenia teoretyczne przeprowadzone dla wiązań wodorowych o średniej mocy wykazały wzrost energii w przeliczeniu na jedno wiązanie wodorowe (rzędu 20%).
– kooperatywność wiązań π - dotyczy sąsiadujących, oddziałujących wzajemnie grup funkcyjnych, posiadających wiązania o charakterze π-elektronowym, połączonych wiązaniami wodorowymi (przepływ ładunku zachodzi poprzez wiązania π). Kooperatywność wiązań π ma istotne znaczenie w strukturach biologicznych, zwłaszcza w białkach i kwasach nukleinowych, gdyż zwiększa własności protonodonorowe i protonoakceptorowe grup C=O i N–H, przyczyniając się do stabilizacji helisy DNA.
Oddziaływania kooperatywne dotyczą nie tylko sąsiadujących, bezpośrednio ze sobą połączonych wiązań wodorowych, ale występują także pomiędzy wiązaniami wodorowymi oddalonymi od siebie.
Tworzenie wiązania wodorowego wiąże się ze zmianami częstości drgań deformacyjnych i rozciągających zarówno grupy protonodonorowej, jak i grupy akceptorowej.
