 Principles of Chemistry
Principles of Chemistry
Roztwory
Roztwory są to jednorodne (homogeniczne) mieszaniny dwóch lub więcej składników, które dzieli się na gazowe, ciekłe lub stałe. Roztwory gazowe otrzymuje się w wyniku zmieszania jednego gazu z drugim. Ponieważ gazy mieszają się ze sobą w dowolnych stosunkach to każda ich mieszanina jest jednorodna, a tym samym jest roztworem. Pod względem kinetycznym roztwór gazowy jest podobny do czystego (jednoskładnikowego) gazu z ta tylko różnicą, że zawiera cząsteczki różnych rodzajów. W stanie doskonałym cząsteczki gazu poruszają się niezależnie od siebie. Roztwory ciekłe otrzymuje się rozpuszczając gaz, ciecz lub ciało stałe w cieczy. Jeżeli cieczą ta jest woda mówimy o roztworach wodnych. W roztworze cząsteczki substancji rozpuszczonej są rozmieszczone bezładnie w całej objętości roztworu. W ujęciu cząsteczkowym trudno mówić o jednorodności ale biorąc pod uwagę, że doświadczenia przeprowadza się na miliardach cząsteczek to roztwór można w takiej skali uważać za jednorodny. Roztworami stałymi są ciała stałe, w których jeden składnik jest rozproszony przypadkowo w stali atomowej lub cząsteczkowej w innym składniku. W czystym krysztale ułożenie atomów jest uporządkowane ale nie dotyczy to sposobu w jaki węzły sieci krystalicznej są zajęte przez te czy inne atomy. Roztwory stałe mają w znacznej mierze postać stopów będących połączeniem dwóch lub więcej pierwiastków mających własności metaliczne. Stop może być związkiem chemicznym lub roztworem. Przykładowo srebro monetarne czy jubilerskie jest roztworem miedzi i srebra. Podobnie mosiądze są stopami (roztworami stałymi) miedzi i cynku. Nie wszystkie stopy są roztworami stałymi; niektóre z nich to niejednorodne mieszaniny złożone z małych kryształów obu składników, a inne mogą być związkami międzymetalicznymi.
Przy omawianiu roztworów operuje się dwoma pojęciami – substancja rozpuszczona i rozpuszczalnik. Substancję występującą w większej ilości określa się zazwyczaj jako rozpuszczalnik, a substancję w mniejszej ilości jako – substancję rozpuszczoną. W niektórych przypadkach pojęcia te można stosować zamiennie. Przykładowo mówiąc o roztworach kwasu siarkowego(VI) i wody kwas określa się jako substancję rozpuszczoną a wodę jako rozpuszczalnik nawet gdy cząsteczki wody w roztworze stanowią mniejszość. Pojęcie solwatowany stosuje się do opisu cząsteczek, które powstają w roztworze w wyniku oddziaływania jednej lub więcej cząsteczek rozpuszczalnika z substancją rozpuszczoną. Jeżeli rozpuszczalnikiem jest woda stosuje się określenie hydratowany (nie solwatowany) lub uwodniony.
Stężenie
Istnieje kilka sposobów określania stężenia roztworów:
ułamek molowy – stosunek liczby moli jednego składnika do sumy liczby moli w roztworze. Np. w roztworze składającym się z 1 mola substancji rozpuszczonej i 3 moli wody ułamek molowy substancji rozpuszczonej wynosi ¼, a ułamek molowy wody ¾. poza ułamkiem molowym stosowane są, choć rzadko, ułamki masowy i objętościowy. Ułamek masowy określa stosunek masy składnika do masy mieszaniny, a ułamek objętościowy stosunek objętości składnika do sumy objętości wszystkich składników przed ich zmieszaniem.
molarność (molowość) – jest to liczba moli substancji rozpuszczonej w 1 dm3 roztworu.
molalność – określa liczbę moli substancji w 1kg rozpuszczalnika.
normalność – określa liczbę gramorównoważników, wyrażoną w gramach, substancji rozpuszczonej w 1 dm3 roztworu.
procent substancji rozpuszczonej – ta forma określania stężenia może odnosić się albo do procentów wagowych lub objętościowych. Procent wagowy określa procent masy substancji rozpuszczonej do całkowitej masy roztworu. Inaczej mówiąc jest to ilość substancji rozpuszczonej zawartej w 100g roztworu. Natomiast procent objętościowy określa objętość substancji rozpuszczonej w 100 cm3 roztworu.
Właściwości roztworów
Interesuje nas jak zmieniają się właściwości rozpuszczalnika po dodaniu do niego substancji rozpuszczonej. Rozpatrzmy następujące doświadczenie. Dwie zlewki, z których jedna zawiera czysty rozpuszczalnik (np. wodę) a druga roztwór jakiejś substancji w tym rozpuszczalniku zostały umieszczone pod kloszem tak jak to przedstawiono na rysunku.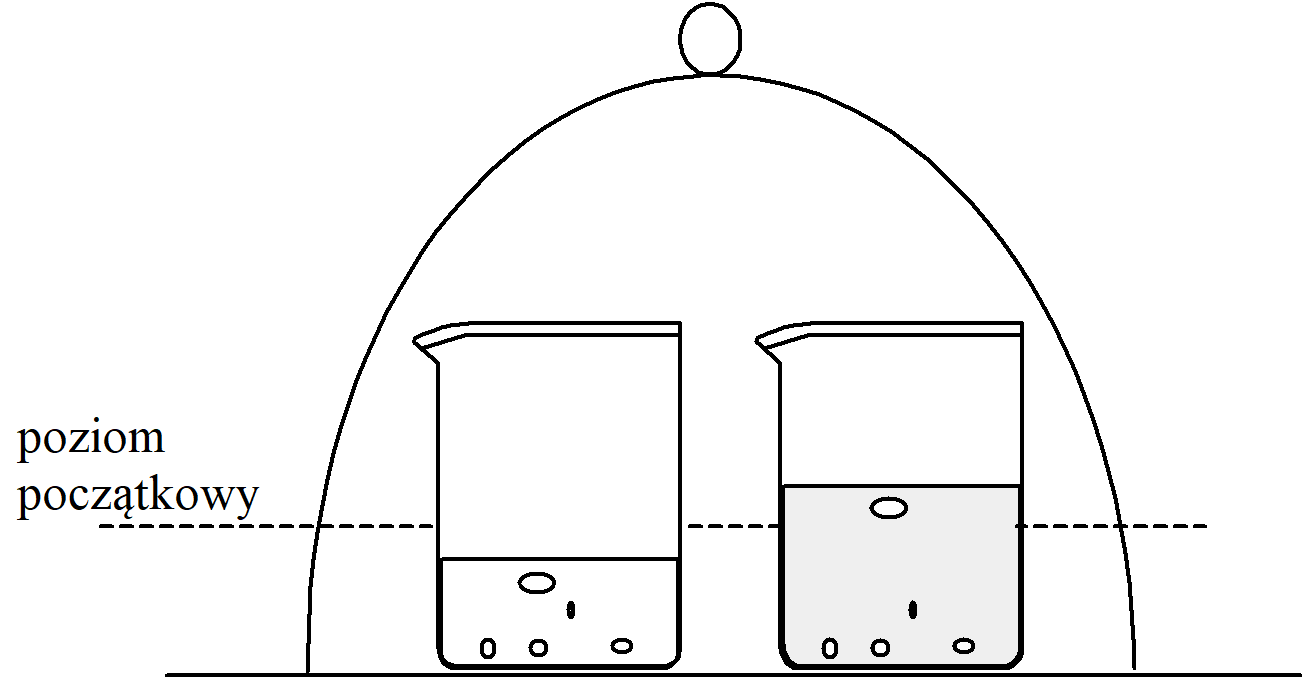 W miarę upływu czasu można zaobserwować, że poziom czystego rozpuszczalnika w zlewce po lewej stronie obniża się, a poziom roztworu w zlewce po stronie prawej podnosi się. Ta obserwacja pozwala na wysnucie wniosku, że zdolność cząsteczek czystego rozpuszczalnika do przechodzenia w stan pary jest większa od zdolności do przechodzenia w fazę gazową rozpuszczalnika z roztworu. Ilościowo zdolność tę opisuje ciśnienie pary. Innym spostrzeżeniem doświadczalnym potwierdzającym pogląd, że dodanie substancji rozpuszczonej zmniejsza zdolność do opuszczania fazy ciekłej przez cząsteczki rozpuszczalnika jest obniżenie temperatury krzepnięcia roztworu. Obniżenie temperatury krzepnięcia i zmniejszenie ciśnienia pary jest w przypadku roztworów rozcieńczonych wprost proporcjonalne do stężenia cząsteczek substancji rozpuszczonej. Obydwa te zjawiska nie zależą, przynajmniej w głównej mierze, od współdziałania między substancją rozpuszczoną a rozpuszczalnikiem . Najważniejszym czynnikiem jest tutaj zmniejszenie stężenia rozpuszczalnika. Jeżeli w roztworze znajduje się substancja rozpuszczona to ilość rozpuszczalnika jest mniejsza od 100%, a tym samym przejście w stan pary jest mniej prawdopodobne niż z czystego rozpuszczalnika. Posługując się pojęciem entropii można powiedzieć, że entropia rozpuszczalnika w roztworze jest większa niż w przypadku czystego rozpuszczalnika. Dodanie substancji rozpuszczonej powoduje zwiększenie prawdopodobieństwa, że cząsteczki rozpuszczalnika pozostaną w fazie skondensowanej.
W miarę upływu czasu można zaobserwować, że poziom czystego rozpuszczalnika w zlewce po lewej stronie obniża się, a poziom roztworu w zlewce po stronie prawej podnosi się. Ta obserwacja pozwala na wysnucie wniosku, że zdolność cząsteczek czystego rozpuszczalnika do przechodzenia w stan pary jest większa od zdolności do przechodzenia w fazę gazową rozpuszczalnika z roztworu. Ilościowo zdolność tę opisuje ciśnienie pary. Innym spostrzeżeniem doświadczalnym potwierdzającym pogląd, że dodanie substancji rozpuszczonej zmniejsza zdolność do opuszczania fazy ciekłej przez cząsteczki rozpuszczalnika jest obniżenie temperatury krzepnięcia roztworu. Obniżenie temperatury krzepnięcia i zmniejszenie ciśnienia pary jest w przypadku roztworów rozcieńczonych wprost proporcjonalne do stężenia cząsteczek substancji rozpuszczonej. Obydwa te zjawiska nie zależą, przynajmniej w głównej mierze, od współdziałania między substancją rozpuszczoną a rozpuszczalnikiem . Najważniejszym czynnikiem jest tutaj zmniejszenie stężenia rozpuszczalnika. Jeżeli w roztworze znajduje się substancja rozpuszczona to ilość rozpuszczalnika jest mniejsza od 100%, a tym samym przejście w stan pary jest mniej prawdopodobne niż z czystego rozpuszczalnika. Posługując się pojęciem entropii można powiedzieć, że entropia rozpuszczalnika w roztworze jest większa niż w przypadku czystego rozpuszczalnika. Dodanie substancji rozpuszczonej powoduje zwiększenie prawdopodobieństwa, że cząsteczki rozpuszczalnika pozostaną w fazie skondensowanej.

Na rysunku obok przedstawiono wykres fazowy, na którym widoczny jest typowy wpływ nielotnej substancji rozpuszczonej o określonym stężeniu na właściwości rozpuszczalnika jakim jest woda. Linie ciągłe przedstawiają wykres fazowy dla czystej wody, a przerywane dla roztworu. Jak widać w przypadku roztworu obszar cieczy jest szerszy tak dla niższych jak i wyższych temperatur w porównaniu do czystego rozpuszczalnika. Jak wynika z rysunku w temperaturze 100°C ciśnienie pary wodnej nad roztworem jest mniejsze od 101 325 Pa (1 atm.) i roztwór nie wrze w tej temperaturze lecz wyższej, w której ciśnienie pary osiągnie odpowiednią wartość. Podobnie wygląda sprawa z temperaturą krzepnięcia, która w roztworze jest niższa niż dla czystego rozpuszczalnika. Obniżenie ciśnienia pary rozpuszczalnika nad roztworem ujmuje prawo Raoulta, według którego częściowe obniżenie ciśnienia pary rozpuszczalnika równa się ułamkowi molowemu substancji rozpuszczonej. Można to zapisać w postaci:
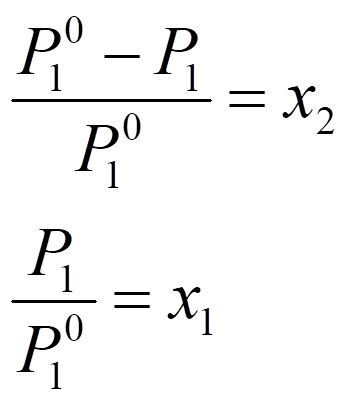
gdzie P10 - ciśnienie pary czystego rozpuszczalnika; P1 ciśnienie cząstkowe rozpuszczalnika nad roztworem; x1 i x2 to ułamki molowe substancji rozpuszczonej i rozpuszczalnika. W rzeczywistości prawo Raoulta jest spełnione jedynie dla roztworów rozcieńczonych.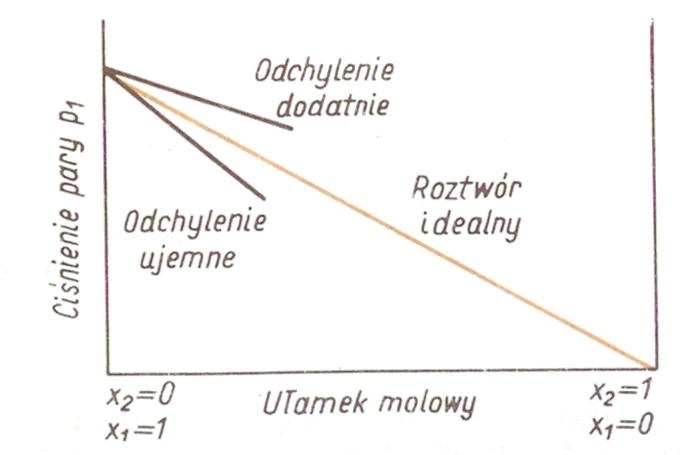 Przy większych stężeniach substancji rozpuszczonej obserwowane są odchylenia dodatnie lub ujemne od zachowania doskonałego.
Przy większych stężeniach substancji rozpuszczonej obserwowane są odchylenia dodatnie lub ujemne od zachowania doskonałego.
Prawo Rauolta określające współzależność między ciśnieniem par i ułamkiem molowym rozpuszczalnika wyjaśnia dlaczego obniżenie temperatury krzepnięcia i podwyższenie temperatury wrzenia jest proporcjonalne do stężenia cząsteczek substancji rozpuszczonej roztworze. Zarówno ze względu na temperaturę krzepnięcia jak i wrzenia stężenie roztworu ma znaczenie dla oznaczenia ułamka molowego rozpuszczalnika. Gdy zwiększa się stężenie roztworu maleje ułamek molowy rozpuszczalnika i maleje proporcjonalnie jego ciśnienie pary. Obniżenie temperatury krzepnięcia i podwyższenie temperatury wrzenia roztworu można zapisać w postaci:
ΔTtk=Kk·m; ΔTtw=Ke·m
gdzie Kk i Ke to odpowiednio stała krioskopowa i ebulioskopowa, m - molalnośc rozpuszczalnika czyli ilość moli substancji rozpuszczonej w 1kg rozpuszczalnika. Wartości stałych krioskopowej i ebulioskopowej nie zależą od substancji rozpuszczonej, a jedynie od rodzaju rozpuszczalnika. Powyższe zależności są spełnione dla roztworów rozcieńczonych, a ponadto substancja rozpuszczona nie może być lotna ani tworzyć roztworu stałego oraz liczba cząsteczek substancji rozpuszczonej musi być równa liczbie cząstek przeniesionych do roztworu. Jeżeli substancja rozpuszczona ulega rozkładowi w roztworze wtedy sumaryczne stężenie wszystkich postaci substancji rozpuszczonej będzie determinowało temperatury krzepnięcia i wrzenia roztworu.
Rozpuszczalność
Termin rozpuszczalność określa jakościowo proces rozpuszczania i jednocześnie jest używany do opisu składu otrzymanych roztworów. Większość roztworów z jakimi mamy do czynienia to roztwory nienasycone, czyli takie do których można dodawać stopniowo substancję rozpuszczoną uzyskując roztwór o innym stężeniu. Każda substancja rozpuszczona i każdy rozpuszczalnik mogą tworzyć wiele roztworów nienasyconych. Jednak w większości przypadków proces dodawania substancji rozpuszczonej nie może być nieskończony. W pewnym momencie osiąga się stan, w którym po dodaniu kolejnej porcji substancji rozpuszczonej do określonej objętości rozpuszczalnika w danej temperaturze nie otrzymuje się roztworu o większym stężeniu gdyż nadmiar dodawanej substancji pozostaje nierozpuszczony. Ilość substancji jako może być rozpuszczona w danej ilości rozpuszczalnika w danej temperaturze jest ograniczona. Roztwór odpowiadający takiemu ograniczeniu nazywa się roztworem nasyconym, a jego stężenie określa się pojęciem rozpuszczalności. Roztwór nasycony pozostaje w stanie równowagi z nadmiarem substancji rozpuszczonej. Równowaga ta jest dynamiczna, gdyż taka sama ilość substancji przechodzi do roztworu jaka jest z niego wydzielana. Roztwór nasycony można zdefiniować jako roztwór, który jest w stanie równowagi z nadmiarem substancji rozpuszczonej. Stężenie roztworu nasyconego, czyli rozpuszczalność, zależy od rodzaju substancji rozpuszczonej,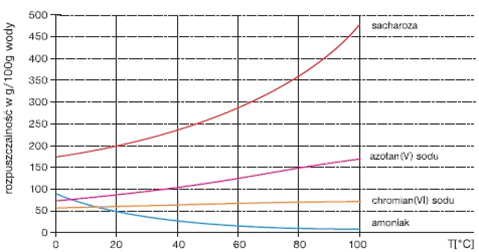 rodzaju rozpuszczalnika, temperatury i ciśnienia. Rozpatrując te zależności należy pamiętać, że w procesie rozpuszczania zachodzą trzy istotne wzajemne oddziaływania: cząsteczki substancji rozpuszczonej oddzielają się od siebie z pobraniem energii; cząsteczki rozpuszczalnika odpychają się od siebie w celu wytworzenia wolnego miejsca na cząsteczki substancji rozpuszczonej co również odbywa się z pobraniem energii; wreszcie cząsteczki rozpuszczalnika przyciągają cząsteczki substancji rozpuszczonej czemu towarzyszy wydzielanie energii. Należy również zwrócić uwagę iż rozpuszczanie substancji skutkuje powstaniem układu o większym stopniu nieuporządkowania niż w przypadku czystego rozpuszczalnika i czystej substancji rozpuszczonej. Zatem rozpuszczanie może zachodzić na skutek wzrostu entropii nawet wtedy gdy energia wzajemnych oddziaływań pomiędzy rozpuszczalnikiem i substancją rozpuszczoną nie jest wystarczająca do skompensowania sumy energii poszczególnych oddziaływań.
rodzaju rozpuszczalnika, temperatury i ciśnienia. Rozpatrując te zależności należy pamiętać, że w procesie rozpuszczania zachodzą trzy istotne wzajemne oddziaływania: cząsteczki substancji rozpuszczonej oddzielają się od siebie z pobraniem energii; cząsteczki rozpuszczalnika odpychają się od siebie w celu wytworzenia wolnego miejsca na cząsteczki substancji rozpuszczonej co również odbywa się z pobraniem energii; wreszcie cząsteczki rozpuszczalnika przyciągają cząsteczki substancji rozpuszczonej czemu towarzyszy wydzielanie energii. Należy również zwrócić uwagę iż rozpuszczanie substancji skutkuje powstaniem układu o większym stopniu nieuporządkowania niż w przypadku czystego rozpuszczalnika i czystej substancji rozpuszczonej. Zatem rozpuszczanie może zachodzić na skutek wzrostu entropii nawet wtedy gdy energia wzajemnych oddziaływań pomiędzy rozpuszczalnikiem i substancją rozpuszczoną nie jest wystarczająca do skompensowania sumy energii poszczególnych oddziaływań.
Rozpatrują zależność rozpuszczalności substancji od rodzaju rozpuszczalnika można stwierdzić, że duża rozpuszczalność występuje wtedy gdy właściwości elektryczne i budowa cząsteczek substancji rozpuszczanej i rozpuszczalnika są podobne. Innymi słowy substancje jonowe jak NaCl dobrze rozpuszcza się w wodzie lecz słabo w rozpuszczalnikach niepolarnych jak benzen. Na rozpuszczalność ma wpływ obecność w roztworze substancji o wspólnym jonie z substancją rozpuszczaną. Rozpuszczalność NaCl w wodzie zawierającej cukier jest niemal taka sama jak w czystej wodzie, jednak rozpuszczalność chlorku sodu w roztworze KCl lub NaNO3 jest znacznie mniejsza od rozpuszczalności w czystej wodzie.
Rozpuszczalność gazów zazwyczaj maleje wraz z temperaturą. Nie jest to jednak regułą gdyż rozpuszczalność niektórych gazów np. helu w wodzie rośnie wraz ze wzrostem temperatury. Podobnie nie ma ogólnej zasady dotyczącej zmian rozpuszczalności cieczy lub ciał stałych wraz ze zmianami temperatury. Zmiany rozpuszczalności wraz z temperaturą są ściśle związane ciepłem rozpuszczania danej substancji, parametrem opisującym zmiany (pobieranie lub emisję) ciepła podczas rozpuszczania substancji w danym rozpuszczalniku. Procesy zachodzące z pobieraniem ciepła z otoczenia nazywa się procesami endotermicznymi , a przebiegające z wydzieleniem ciepła to procesy egzotermiczne. To czy rozpuszczanie jest procesem endo- czy egzotermicznym zależy od szeregu czynników, między innymi rodzaju substancji rozpuszczonej i rozpuszczalnika jak również od względnych wartości dwóch energii: energii potrzebnej do rozerwania sieci krystalicznej ciała stałego oraz energii wydzielanej podczas solwatacji cząsteczek substancji rozpuszczonej. Rozpatrując termodynamiczne efekty rozpuszczania można stwierdzić, że jeżeli mamy roztwór nasycony dla którego zmiana entalpii ΔH w procesie:
ciało rozpuszczone + rozpuszczalnik → roztwór
jest dodatnie (reakcja endotermiczna) rozpuszczalność zwiększa się wraz ze wzrostem temperatury. Natomiast w przypadku gdy ΔH jest ujemne (proces egzotermiczny) wzrost temperatury powoduje obniżenie rozpuszczalności. Te efekty ujmuje zasada Le Châteliera. Jeżeli w procesie rozpuszczania ciepło jest pochłaniane to działanie bodźca zakłócającego stan równowagi procesu wskutek podwyższenia temperatury może być złagodzone przez intensywne rozpuszczanie. Jeżeli w procesie rozpuszczania wydziela się ciepło to działanie bodźca wywołanego podwyższeniem temperatury może być złagodzone przez wytrącanie substancji rozpuszczonej.
Wpływ ciśnienia najłatwiej zaobserwować na przykładzie rozpuszczania gazów, których rozpuszczalność wzrasta wraz ze wzrostem ciśnienia gazu nad roztworem. Ilościowo proces ten opisuje prawo Henry’ego, zgodnie z którym w stałej temperaturze ułamek molowy gazu rozpuszczanego w rozpuszczalniku jest wprost proporcjonalny do jego ciśnienia cząstkowego nad rozpuszczalnikiem. Prawo to ma postać:
K=P/x
gdzie K - stała Henry’ego, P - ciśnienie cząstkowe ciała rozpuszczonego w fazie gazowej, x - ułamek molowy gazu w roztworze.
W przypadku cieczy i ciał stałych rozpuszczalność zasadniczo nie zależy od ciśnienia. Zmiany objętości roztworów wraz ze zmianami ciśnienia są na tyle małe, że dopiero ciśnienia rzędu tysięcy atmosfer mogą wywoływać zmiany w rozpuszczalności.
Należy jeszcze na koniec nadmienić, że istnieje możliwość otrzymania roztworu wykazującego większe stężenie substancji rozpuszczonej niż w roztworze nasyconym. Roztwory akie nazywane są roztworami przesyconymi i są one nietrwałe gdyż łatwo wydzielają nadmiar substancji rozpuszczonej. Z reguły roztwory takie można otrzymać przez powolne schładzanie roztworu nasyconego w podwyższonej temperaturze. Po ochłodzeniu jeżeli roztwór nie zostanie poddany działaniu bodźców zewnętrznych np. mechanicznych jak uderzenie, wstrząs czy odrobina pyłu wpadająca do roztworu, czy nawet zadrapanie ścianki naczynia, jest roztworem przesyconym, w którym stężenie substancji rozpuszczonej w danej temperaturze jest większe niż w roztworze nasyconym.
Koloidy
Omawiając roztwory przyjęliśmy że łatwo jest odróżnić mieszaniny jednorodne od niejednorodnych. Istnieją roztwory, które nie są w sposób oczywisty jednorodne lub niejednorodne. Roztwory takie noszą nazwę koloidów. Cząstki rozproszone w układzie koloidalnym są na tyle małe, że nie tworzą w sposób oczywisty odrębnej fazy, ale jednocześnie nie na tyle małe aby można było mówić o rozworze rzeczywistym. Z roztworu koloidalnego pozostawionego przez dłuższy czas cząstki koloidalne nie wydzielają się ze znaczną szybkością, są niewidoczne pod mikroskopem i nie można ich oddzielić przez sączanie. Zazwyczaj układy koloidalne określane są za pomocą rozmiaru cząstek. Jeżeli wielkość cząstek zawiera się w granicach od 10-7 do 10-4 cm to mówimy o roztworze koloidalnym. Wielkość cząstek rozproszonych w układzie koloidalnym nie świadczy o ich budowie, mogą to być atomy, pojedyncze cząsteczki lub grupy atomów, cząsteczek jak i pojedyncze duże cząsteczki. Koloidy często klasyfikuje się na podstawie stanu skupienia faz z jakich są złożone. Generalnie koloid składa się z: fazy ciągłej, czyli substancji rozpraszającej, zwanej też ośrodkiem dyspersyjnym lub dyspergującym oraz fazy rozproszonej, czyli substancji zdyspergowanej w ośrodku dyspersyjnym i w nim nierozpuszczalnej. W najważniejszym podziale koloidów wyróżnia się zole, emulsje, żele i aerozole. W zolach ciało stałe jest rozproszone w cieczy. Emulsje są koloidami, w których ciecz jest rozproszona w cieczy. Żele złożone są cieczy, która zawiera ciało stałe tworzące sieć przenikającą cały układ. Obie fazy, stała i ciekła, są fazami ciągłymi. Aerozol to koloid powstający w wyniku rozproszenia ciała stałego lub cieczy w gazie.
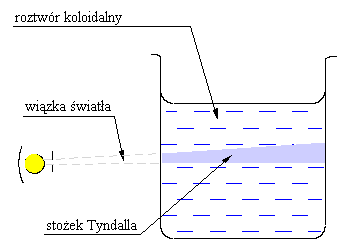 Jeżeli wiązka światła jest przepuszczana przez roztwór rzeczywisty lub czysty rozpuszczalnik to patrząc z boku nie można jej dostrzec. Jeżeli jednak wiązka światła pada na roztwór koloidalny to można ją zaobserwować z boku. Efekt ten jest nazywany efektem Tyndalla. Zjawisko związane jest z uginaniem się promieni na cząstkach fazy rozproszonej. Intensywność tego zjawiska jest tym większa, im większa jest różnica między współczynnikiem załamania fazy rozproszonej i ośrodka dyspersyjnego. Zależy również od długości rozpraszanej fali – silniej rozpraszane są krótsze. Obserwacja stożka Tyndalla pod mikroskopem oczywiście nie pozwala na zauważenie cząstek koloidalnych, ale ich położenie daje się określić na podstawie pojawiania się punktów światła. Obserwowane cząstki podlegają ruchom Browna. Gdy koloid jest przechowywany w nieizolowanym pojemniku to nie wykazuje on skłonności do rozdzielania się faz. Natomiast w koloidzie w dobrze izolowanym pojemniku po pewnym czasie następuje rozdzielenie faz. Jest to spowodowane działaniem dwóch przeciwstawnych czynników jakimi są siła przyciągania ziemskiego powodująca opadanie cięższych cząstek (faza rozporoszona) oraz rozpraszaniem pod wpływem ruchów Browna. W przypadku koloidu znajdującego się w nieizolowanym pojemniku prądy konwekcyjne wywoływane przez różnice temperatury w poszczególnych miejscach roztworu zapobiegają separacji faz.
Jeżeli wiązka światła jest przepuszczana przez roztwór rzeczywisty lub czysty rozpuszczalnik to patrząc z boku nie można jej dostrzec. Jeżeli jednak wiązka światła pada na roztwór koloidalny to można ją zaobserwować z boku. Efekt ten jest nazywany efektem Tyndalla. Zjawisko związane jest z uginaniem się promieni na cząstkach fazy rozproszonej. Intensywność tego zjawiska jest tym większa, im większa jest różnica między współczynnikiem załamania fazy rozproszonej i ośrodka dyspersyjnego. Zależy również od długości rozpraszanej fali – silniej rozpraszane są krótsze. Obserwacja stożka Tyndalla pod mikroskopem oczywiście nie pozwala na zauważenie cząstek koloidalnych, ale ich położenie daje się określić na podstawie pojawiania się punktów światła. Obserwowane cząstki podlegają ruchom Browna. Gdy koloid jest przechowywany w nieizolowanym pojemniku to nie wykazuje on skłonności do rozdzielania się faz. Natomiast w koloidzie w dobrze izolowanym pojemniku po pewnym czasie następuje rozdzielenie faz. Jest to spowodowane działaniem dwóch przeciwstawnych czynników jakimi są siła przyciągania ziemskiego powodująca opadanie cięższych cząstek (faza rozporoszona) oraz rozpraszaniem pod wpływem ruchów Browna. W przypadku koloidu znajdującego się w nieizolowanym pojemniku prądy konwekcyjne wywoływane przez różnice temperatury w poszczególnych miejscach roztworu zapobiegają separacji faz.
Układy koloidalne są termodynamicznie nietrwałe w odniesieniu do fazy zwartej. Niestabilność te można wyrazić termodynamicznie zauważając, że zmiana entalpii swobodnej dG, zachodząca gdy powierzchnia próbki zmienia się o dσ w stałej temperaturze i pod stałym ciśnieniem, wynosi dG = γdσ, gdzie γ jest napięciem międzyfazowym. Wynika stąd, że dG < 0 jeśli dσ < 0. Trwałość koloidów musi być więc związana z kinetyką ich rozpadu; koloidy są niestabilne termodynamicznie, ale kinetycznie są nielabilne. Na pierwszy rzut oka argument kinetyczny nie wydaje się spełniony, gdyż nawet znacznie od siebie oddalone cząstki koloidalne przyciągają się i te oddziaływania dalekiego zasięgu wywołują tendencję do skupiania się razem. Fakt ten wyjaśnia następujące rozumowanie. Energia przyciągania dwóch pojedynczych atomów i oraz j, odległych o Rij w jednej cząstce koloidu zmienia się jako funkcja 1/R6ij. Jednakże suma wszystkich oddziaływań między parami atomów maleje z odległością jak 1/R2., gdzie R jest odległością między środkami cząstek. Ta suma ma znacznie dalszy zasięg niż przewiduje to zależność typu 1/R6 charakterystyczna dla pojedynczych i małych cząstek. Dalej istnieją czynniki, które przeciwstawiają się przyciąganiom dalekozasięgowym. Na powierzchni cząstek koloidu tworzy się warstewka ochronna, która stabilizuje granicę faz.
Średnie masy molowe makrocząsteczek
Polimery mogą występować w formie mono– lub polidyspersyjnej w tym znaczeniu, że próbka jest mieszaniną cząsteczek o różnych długościach łańcucha i różnych masach molowych dając w rezultacie różne typy wartości średnich charakteryzujących układ. Wartość średnia otrzymana na podstawie pomiarów osmometrycznych daje liczbowo średnią masę molową, która stanowi wartość uzyskaną przez ważenie każdej masy molowej liczbą cząstek N, reprezentujących daną masę molową Mi w próbce:
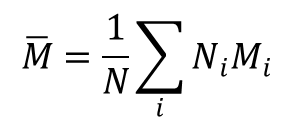
gdzie Ni jest liczbą cząstek o masie molowej Mi, a N oznacza liczbę wszystkich cząstek w próbce. W zależności od metody jaką wyznacza się średnią masę molową otrzymujemy lepkościową średnią masę molową gdy mierzy się lepkość roztworu. Pomiary rozpraszania światła pozwalają na wyznaczenie masowo średniej masy molowej, a pomiary czasu sedymentacji tzw. z-średnią masę molową. Różne wartości tak wyznaczonych średnich mas molowych pozwalają na wyznaczenie współczynnika heterogeniczności próbki (współczynnika polidyspersji). Współczynnik ten wyznacza się jako iloraz średnich mas molowych wyznaczonych na podstawie pomiarów lepkościowych i sedymentacyjnych. Jeżeli współczynnik heterogeniczności próbki jest mniejszy niż 1,1 (polimery syntetyczne)to o takim polimerze mówi się, że jest monodyspersyjny.
Właściwości koligatywne makrocząsteczek
Makrocząsteczki tworzą roztwory znacznie odbiegające od stanu idealnego z uwagi na to, że ich cząsteczki są duże przez co wypierają znaczne ilości rozpuszczalnika. W takim wypadku szczególnie istotna staje się zmiana entropii związana z rozpuszczaniem makrocząsteczki. Duże rozmiary makrocząsteczek powodują, że nie są one zdolne do swobodnego ruchu w roztworze, gdyż są wyłączone z obszarów zajmowanych przez inne cząsteczki substancji rozpuszczonej. Dla roztworów makrocząsteczek obserwuje się znaczne udziały ich entalpii w entalpii swobodnej roztworu co jest związane z tym, że oddziaływanie rozpuszczalnik rozpuszczalnik jest bardziej uprzywilejowane niż oddziaływanie makrocząsteczka–rozpuszczalnik. Objętość wyłączona wnosi wkład do nadmiarowej entropii roztworu, a oddziaływania przyciągające i odpychające między makrocząsteczkami dają wkład do nadmiarowej entalpii. Większość układów typu rozpuszczalnik- substancja rozpuszczona charakteryzuje się określoną temperaturą, w której te efekty się znoszą dają roztwór pozornie idealny. W przypadku roztworów makrocząsteczek temperatura ta nosi nazwę temperatury theta Flury’ego. W temperaturze θ wiralny współczynnik osmotyczny (współczynnik przy drugim członie w równaniu van’t Hoffa na ciśnienie osmotyczne: π=[X]RT(1+B[X] + C{X] + ...)) jest równy zeru. W kategoriach cząsteczkowych można przyjąć, że w temperaturze θ cząsteczki znajdują się w stanie niezaburzonym.
Konformacja i konfiguracja makrocząsteczek
Struktura pierwszorzędowa jest określona przez sekwencję małych jednostek konstytucyjnych tworzących sieć. W przypadku jednorodnych polimerów syntetycznych są to cząsteczki monomeru. W przypadku makrocząsteczek kopolimerów i polimerów biologicznych określenie struktury pierwszorzędowej jest trudniejsze i nierzadko wymaga stosowania technik sekwencjonowania.
Struktura drugorzędowa odnosi się do, często lokalnie, dobrze scharakteryzowanego uporządkowania jednostek strukturalnych. Proces niszczenia struktury drugorzędowej nosi nazwę denaturacji. Różnica pomiędzy strukturą pierwszo– i drugorzędową jest ściśle związana z różnicą pomiędzy konfiguracją i konformacją łańcucha polimeru. Określenie konfiguracja odnosi się do tych cech strukturalnych, które mogą być zmienione jedynie przez rozerwanie wiązań chemicznych i utworzenie nowych; łańcuchy –A–B–C– i –A–C–B– mają różne konfiguracje. Termin konformacja dotyczy przestrzennego uporządkowania różnych części łańcucha; z jednej konformacji można przejść do drugiej przez rotację jednej części łańcucha wokół wiązania.
Struktura trzeciorzędowa odpowiada ogólnej trójwymiarowej strukturze cząsteczki.
Struktura czwartorzędowa dotyczy sposoby w jaki niektóre cząsteczki tworzą się w wyniku agregacji.